How to Automate Validation Documentation
Automating validation documentation sounds either obvious or reckless depending on who you ask. The engineer drowning in test-step tables wants it yesterday. The quality lead who owns the signature worries that automation and data integrity do not mix. Both are right about something. The work is genuinely automatable, and the line between what you can automate and what you cannot is the whole point.
This post draws that line clearly. It covers what parts of validation documentation can be automated safely, what has to stay with a qualified human, how a defensible automated workflow is put together, and how to keep 21 CFR Part 11 and data integrity intact while you do it. The goal is a workflow that removes the repetitive drafting burden without ever removing the accountability.
If you want to try the drafting side directly, you can generate a structured protocol from your equipment context and see where automation ends and review begins. Start at /signup, or read the model first.
What "automating validation documentation" actually means
Validation documentation is not one task. It is a chain: capturing the equipment or process context, deciding scope and risk, writing test steps and acceptance criteria, building traceability, executing the protocol and capturing evidence, reviewing, approving, and maintaining the controlled record over time. Some links in that chain are repetitive and rule-bound. Others require professional judgment and a signature that carries legal accountability.
Automation belongs on the repetitive, rule-bound links. It does not belong on the judgment-and-accountability links. Confusing the two is where automation goes wrong, either by leaving obvious drafting toil in place or by pretending a machine can own a decision a qualified person is required to own.
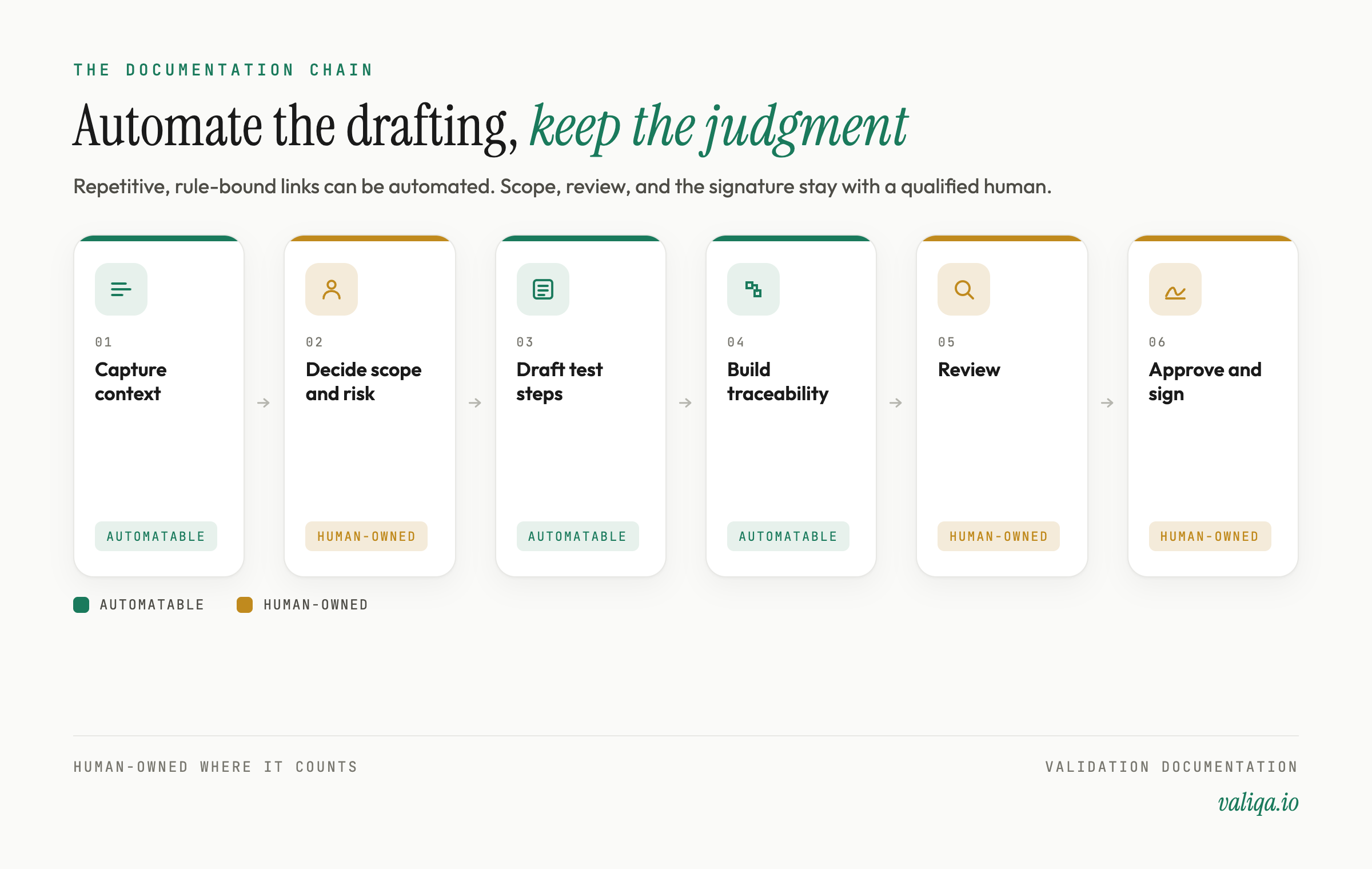
What you can automate safely
Structure and formatting. Section layout, numbering, the approval block, the traceability table, deviation-handling boilerplate. This is identical across protocols and is exactly where inconsistency causes findings. Automate it fully. There is no judgment in a section number.
First-draft test steps and acceptance criteria. Given a complete equipment context and the relevant specifications, a tool can draft measurable, sourced test steps and acceptance criteria far faster than hand-authoring. This is a draft, not a final. It goes to review. But drafting is the repetitive part, and drafting is automatable.
Traceability matrix construction. Mapping each requirement to a test and a result is mechanical once the requirements and tests exist. Building the matrix alongside the test steps, automatically, turns it from an end-of-project cleanup into a live coverage check. See the traceability matrix in validation.
Regulatory mapping scaffolding. Pulling the right predicate regulation, consensus standard, and guidance references into the right sections, quality-system protocols to 21 CFR Part 820, pharma to Part 210/211, Part 11 where electronic records apply, is scaffolding a tool can assemble for a human to confirm.
Export and packaging. Producing a clean, audit-ready PDF or Word file with the matrix, evidence sections, and approval block laid out correctly. This is pure mechanics and should never be manual.
What you cannot automate, and should not try to
The risk decisions. Which attributes are critical, how deep to test, what to accept and what to reject. A tool can ingest your risk assessment and scope a draft against it. It cannot own the risk judgment. That stays with your validation engineer and your quality process.
Review and approval. A qualified human has to review the draft, confirm the tests are right, confirm the criteria are sourced, and approve it. Regulators expect a qualified person to sign their name, and that signature carries professional and legal accountability. Automation produces the draft the human reviews. It does not become the reviewer.
Execution judgment and deviation disposition. When an executed step falls out of range, deciding whether it is a true deviation, what caused it, and how to disposition it is judgment work. A system can capture the record and route it. It cannot make the call.
Final accountability for the content. The protocol is a professional attestation about a specific piece of equipment or process. The person who signs it owns whether it is correct. No automation transfers that ownership.
This boundary is not a limitation to work around. It is the design. A workflow that respects it is faster and defensible. A workflow that violates it is faster and indefensible, which is to say worthless the first time an inspector reads it.
How CSA changes the automation conversation
FDA's Computer Software Assurance approach, now final guidance, is directly relevant here, and not for the reason people assume. CSA is a risk-based approach to assuring the software you use in production and quality systems: focus assurance effort where the software's failure would affect product quality or data integrity, and use lighter, more efficient evidence where risk is low. We break it down in CSA vs CSV: what FDA's finalized guidance actually changed.
The connection to automating documentation is twofold. First, CSA is itself an argument for less documentation toil where risk is low, which is the whole premise of automating the repetitive links. Second, if you use software to help produce validation documentation, that software sits inside your assurance thinking. A tool that produces drafts a human reviews and approves is lower risk than one making unreviewed decisions, and the CSA lens makes that distinction explicit. The GAMP 5 categories, covered in GAMP 5 categories explained, are the other half of that framing.
Keeping Part 11 and data integrity intact
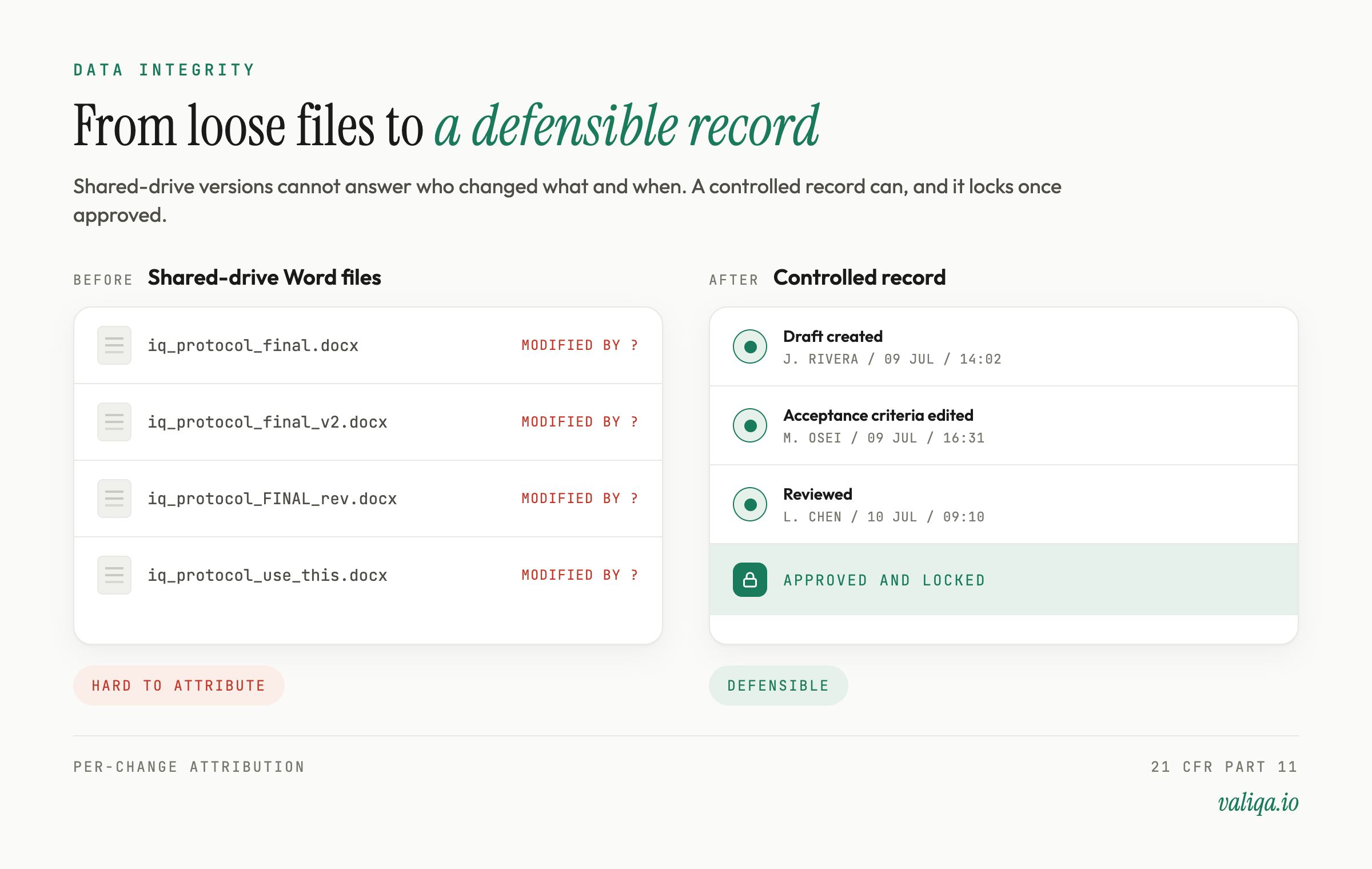
Automation raises the stakes on records because a machine can generate and modify records quickly. That makes the controls more important, not less.
Every automated draft and every human edit should be captured with the user, a timestamp, and a before-and-after record. Approved protocols should be locked from silent edits and changed only through a controlled revision. Electronic signatures should be controlled in line with 21 CFR Part 11 where they apply, with the procedural and predicate-rule controls behind them. And the records the tool produces should satisfy ALCOA+: attributable, legible, contemporaneous, original, accurate, complete, consistent, enduring, and available. We cover the electronic-records layer in how to write a Part 11 compliant CSV protocol.
Done right, automation improves data integrity rather than threatening it, because a well-built tool enforces attribution and versioning that a shared drive of Word files never will.
A workflow that automates the toil and keeps the judgment
Put it together and the defensible automated workflow looks like this. Capture the equipment or process context once, completely. Let the tool draft the structured protocol, test steps, acceptance criteria, traceability matrix, and regulatory scaffolding. Have your qualified engineer review the draft against a fixed checklist, adjust the risk scope and criteria using their judgment, and approve it inside your quality system. Export the final audit-ready document in one clean step. Maintain the controlled record with full change history.
The repetitive links are automated. The judgment and signature links stay human. That is the shape of validation documentation automation that actually holds up.
Valiqa is built to that shape. You enter the equipment context, and you get a structured protocol draft with measurable acceptance criteria, a built-in traceability matrix, regulatory scaffolding, and an audit-ready export, with full change tracking. It is a draft your qualified engineer reviews and approves. It automates the drafting toil and never touches the accountability. The self-serve model with transparent pricing means a mid-market team without a validation department can adopt it without a procurement cycle: see /pricing, start at /signup, or pick your protocol type with the protocol selector.
If you want to know whether an automation tool is even the right move for your team versus a hardened template and a checklist, the honest self-scoring at /evaluate will tell you.
Frequently Asked Questions
Ready to automate your validation documentation?
Generate audit-ready IQ/OQ/PQ protocols in minutes, not weeks.
Get StartedWe use essential cookies for authentication and security. With your consent, we also use Microsoft Clarity on our marketing pages to understand how visitors navigate the site. Learn more.