CSA vs CSV: What FDA's Final Guidance Changed
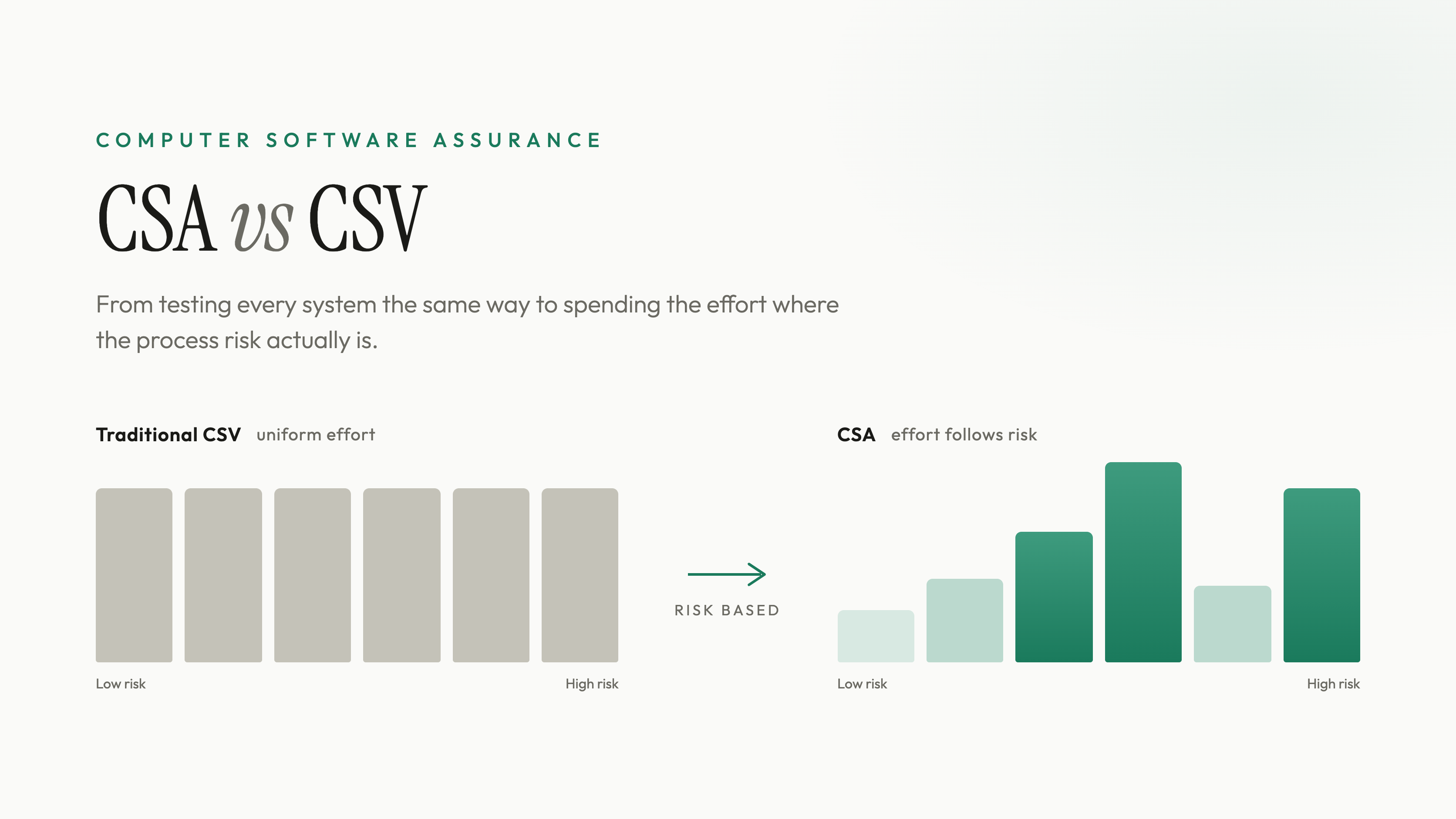
If you have ever validated a piece of production software the old way, you know the feeling. A boxed historian, used exactly as it ships, gets the same thick scripted protocol as the custom batch-disposition logic you wrote from scratch. Every screen gets a screenshot. Every field gets a test case. The binder gets heavier, the schedule slips, and at the end of it the highest-risk software in the room got no more scrutiny than the lowest-risk software, because the process treated them the same. That outcome was never what the regulations asked for. It was what computer system validation drifted into.
Computer Software Assurance, CSA, is the FDA telling you to stop doing that. It is now final guidance, not a draft, and it was revised in early 2026 to line up with the new Quality Management System Regulation. This guide is for the engineer who actually configures the SCADA system and writes the PLC logic, not the person who files the binder. It covers what CSV was and where it went wrong, what CSA actually is and where it sits relative to the regulations, what genuinely changed between the two, what did not change so you do not overcorrect, how the 2026 QMSR revision fits, a worked example on a production system, and the misconceptions that cause teams to get CSA wrong in both directions.
What computer system validation actually was
Computer system validation is the traditional practice of establishing documented evidence that a computerized system does what it is supposed to do and will keep doing it. The idea itself is sound and still required. The regulations have always expected that software used in production and the quality system is fit for purpose and under control. CSV was the industry's standard way of demonstrating that, built around scripted protocols, predefined expected results, and a documented record of every executed step.
The problem was never the concept. It was the way CSV calcified in practice. Over time, "validate the software" quietly turned into "produce the maximum possible documentation for every system, regardless of what the system does or what happens if it fails." Teams wrote exhaustive scripted test cases for low-risk, off-the-shelf features that the vendor had already tested extensively. Screenshots became the deliverable instead of the evidence. Effort got measured in binder thickness rather than in risk retired. And because the same heavy process applied to everything, the genuinely risky software, the custom code that could actually corrupt a batch record or release a bad product, did not get proportionally more attention. It got the same attention as the screen-formatting feature next to it.
That is the failure mode CSA was written to correct. Not validation itself, but the documentation-for-its-own-sake culture that grew up around it and the false sense of control it produced.
What Computer Software Assurance actually is
Computer Software Assurance is FDA guidance on how to apply a risk-based, least-burdensome approach to the software used in medical device production and the quality system. The FDA issued it in draft in September 2022, finalized it on September 24, 2025 as "Computer Software Assurance for Production and Quality System Software," and revised it on February 3, 2026 as "Computer Software Assurance for Production and Quality Management System Software" to align with the QMSR. It is final, current guidance. Anyone still describing CSA as a draft is working from an outdated picture.
Two things about its nature matter before anything else. First, CSA is guidance, not a regulation. Like GAMP 5, it does not carry independent legal force. It describes the FDA's recommended way to satisfy software-validation expectations that already exist under the QMSR, and it intersects with 21 CFR Part 11 where the system handles electronic records or electronic signatures that fall within Part 11 scope. When CSA was finalized in 2025 it superseded Section 6 of the FDA's older General Principles of Software Validation guidance, making the risk-based assurance model the agency's recommended framework for this software. Second, CSA has a specific scope. It applies to software used as part of device production or the quality system: the SCADA, MES, LIMS, historians, PLC logic, automated inspection, and quality-system tools that run and govern manufacturing. It does not cover device software functions such as software as a medical device or software embedded in a medical device, which follow a separate validation pathway.
CSA does not lower the bar. It moves the bar to where the risk is. The whole point is to take the effort that was being spread evenly across everything and concentrate it on the software whose failure could actually compromise product quality, data integrity, or patient safety, while letting the low-risk software earn a lighter, faster touch.
What actually changed
The shift from CSV practice to CSA is a change of starting point, and everything else follows from it. Traditional CSV started with the document set: here is the protocol template, fill it in for every system. CSA starts with two linked questions: what is the intended use of this specific software feature, function, or operation, and what process risk follows if it fails?
Once you answer those questions first, the assurance activity falls out of it. High process risk, especially where a failure could also pose a medical device risk, earns rigorous scripted testing with predefined steps and expected results. Lower process risk earns lighter, faster methods. That single reordering, risk before documentation instead of documentation regardless of risk, is the core of what changed.

| Dimension | Traditional CSV in practice | Computer Software Assurance |
|---|---|---|
| Starting point | Documentation template applied to every system | Intended use first, then process risk for each feature |
| Test rigor | Uniform scripted protocols regardless of risk | Rigor scaled to risk: scripted for high, unscripted for low |
| Low-risk features | Full scripted test cases and screenshots | Unscripted testing: scenario, exploratory, error-guessing |
| Vendor testing | Often re-proven from scratch | Leveraged as objective evidence where justified |
| Documentation | Volume as the goal | Right-sized objective evidence proportionate to risk |
| Mindset | Validation as a paperwork exercise | Assurance as a confidence-building activity |
The second change is the explicit blessing of unscripted testing. Under CSA, software features that are not high process risk can be assured with unscripted methods: scenario testing, exploratory testing, error-guessing, ad-hoc testing, or a combination suited to the risk. You do not need a predefined script and a screenshot for every interaction with a low-risk function. You need enough assurance, recorded as enough objective evidence, to be confident the software does what you need. Scripted testing does not disappear. It gets reserved for the high-risk software where the structured rigor actually buys you something.
The third change is leverage. CSA explicitly encourages using supplier and developer activities as objective evidence rather than re-testing what a competent vendor already tested. If a vendor validated a platform feature and you can justify relying on that evidence, you do not automatically re-perform all of that work. You still test what is specific to your intended use, configuration, interfaces, and risk. This is the same instinct the GAMP 5 category system builds in: higher-category effort buys you the right to inherit lower-category work from a competent supplier.
The fourth change is the redefinition of the deliverable. The output of CSA is not a binder. It is the right amount of objective evidence to support a justified conclusion that the software is fit for its intended use. Sometimes that evidence is a robust scripted protocol. Sometimes it is a short record of an exploratory test session, an electronic log, or a documented review of vendor evidence. The format follows the risk, and the screenshot reflex that defined late-stage CSV is exactly what CSA is telling you to drop where it adds no assurance.
What did not change
CSA is widely misread as permission to do less. That reading gets teams in trouble, so it is worth being precise about what did not move.
The legal requirement to have assured, fit-for-purpose software did not change. Under the QMSR, production and quality-system software still has to be validated for its intended use and kept under control. Where that software also creates, modifies, maintains, archives, retrieves, or transmits electronic records or signatures within Part 11 scope, Part 11 controls still apply, and a Part 11 compliant CSV protocol is where you demonstrate them. CSA changes how you demonstrate that, not whether you have to.
Data integrity and electronic-records requirements did not change. 21 CFR Part 11 still governs electronic records and electronic signatures, and the ALCOA+ expectations for attributable, legible, contemporaneous, original, and accurate records still apply to the data your systems produce. A lighter test approach for a low-risk feature does not loosen the data-integrity controls on the records that feature touches.
The rigor on high-risk software did not drop. For the custom logic that gates batch disposition or controls a critical process parameter, CSA expects more focused, rigorous testing, not less. CSA is a reallocation of effort, not a reduction of it. The hours you save on the boxed historian are hours you are meant to spend on the bespoke routine that can actually hurt the product.
And change control did not change. When production software is modified, you still assess the change and re-establish assurance proportionate to its risk, the same judgment that governs whether a software update triggers revalidation. CSA tells you to scale that re-assurance to risk. It does not tell you to skip it.
How the 2026 QMSR revision fits
The reason CSA has two final versions in the space of five months is the QMSR. The FDA Quality Management System Regulation took effect on February 2, 2026 and restructured 21 CFR Part 820 to incorporate ISO 13485:2016 by reference. The September 2025 CSA guidance was written against the older Quality System Regulation vocabulary. The February 3, 2026 revision re-aligned it, which is why the title changed from "Quality System Software" to "Quality Management System Software" and the references were updated to the QMSR framework.
For the engineer, the practical effect is in where the software-assurance expectation now lives. Under the QMSR, the relevant anchor is ISO 13485:2016, which spreads software validation across three clauses rather than one: clause 4.1.6 covers software used in the quality management system, clause 7.5.6 covers software used in production and service provision where the output cannot be fully verified by later inspection, and clause 7.6 covers software used to monitor and measure. Cite the clause that matches the software's actual use rather than collapsing everything into one. Teams whose documentation predates the transition will still see the pre-QMSR anchor for automated process software at 21 CFR 820.70(i), and that historical reference is fine in legacy records as long as current work uses the QMSR framing.
None of this changed the substance of CSA. The risk-based, least-burdensome approach the FDA introduced in the 2022 draft carried straight through both final versions. The 2026 revision is an alignment of terminology and references to current law, not a new direction.
A worked example: applying CSA on a production line
Concrete beats abstract, so take the same kind of system the GAMP 5 guide decomposed: a packaging line with a PLC handling machine logic, a SCADA application for the operator interface, a vision unit checking label placement, and a historian capturing batch data. GAMP 5 tells you how much of each component you built versus inherited. CSA tells you how to assure each one based on what happens if it fails. The two questions stack.
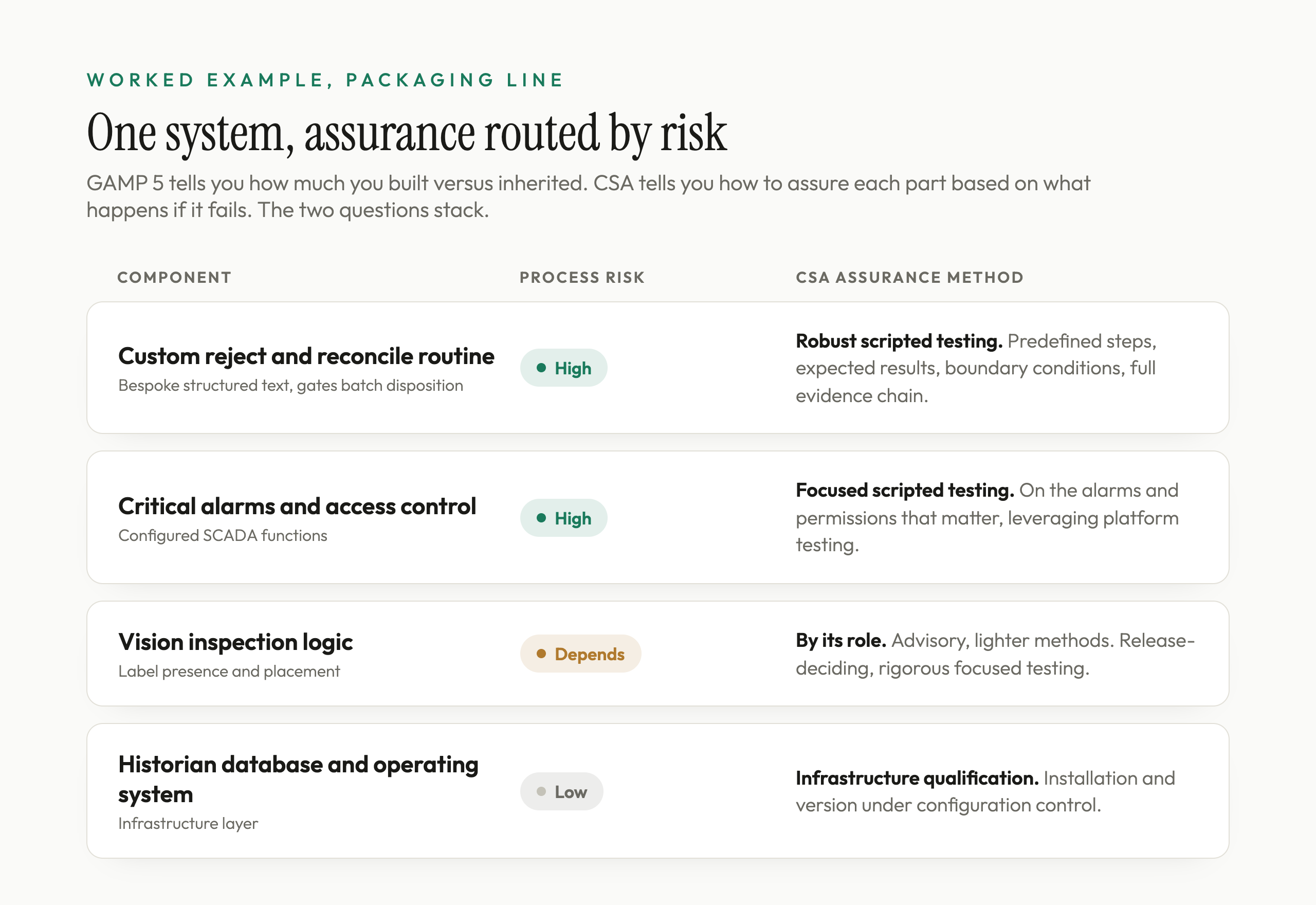
Start with process risk, feature by feature. The custom structured-text routine that counts rejects, reconciles them against the batch count, and holds the batch if reconciliation fails is high process risk. Its failure could release an unreconciled batch, a direct product-quality and patient-safety concern. CSA routes rigorous scripted testing here: predefined steps, expected results, boundary conditions, and a documented evidence chain, exactly because this is where a software failure does real harm. This is also the Category 5 custom code from the GAMP analysis, so the high-effort assurance and the high-category lifecycle land on the same component, which is the point.
The SCADA alarm and access-control configuration is moderate to high process risk depending on what those alarms protect. Assure it with focused scripted testing on the alarms and permissions that matter, and lean on the vendor's platform testing for the underlying SCADA engine rather than re-proving it.
The vision unit can land on either side of the line depending on intended use. If it only provides advisory information and a separate control prevents mislabeled product from being released, some of its functions are not high process risk and can be assured with lighter methods plus supplier evidence where justified. If it is the configured control that decides accept or reject for label presence or placement, its decision logic is high process risk and warrants rigorous, focused testing of that logic. The point is that the testing follows the function's role in the process, not the fact that the box came from a vendor.
The historian's underlying database and the server operating system are low process risk as infrastructure. You confirm installation and version under configuration control and manage them through infrastructure qualification, not a separate full scripted functional protocol.
Notice what CSA did. It took the same finite number of validation hours and pushed them toward the features where a software failure reaches the product, the custom reject routine, the critical alarms, and any inspection logic that gates release, and pulled them away from the infrastructure and the off-the-shelf functions that cannot, like the historian's database engine and the operating system. That is the entire idea: assurance proportionate to risk, recorded as objective evidence proportionate to risk, with the scripted rigor spent where it earns its cost.
Common misconceptions that get teams in trouble
The first misconception is that CSA means less validation. It does not. It means right-sized assurance. For low-risk software you do less, and for high-risk software you often do more and more focused work. A team that reads CSA as a blanket license to cut testing has misread it and will be exposed exactly where it matters, on the high-risk software CSA wanted them to scrutinize harder.
The second is that CSA means no documentation. CSA still requires objective evidence. What changed is that the evidence is proportionate to risk and is not padded with screenshots that prove nothing. An exploratory test session still gets recorded. A reliance on vendor evidence still gets justified and documented. Less ceremony is not no record.
The third is calling CSA a draft or a 2022 proposal. CSA is final guidance, finalized September 24, 2025 and revised February 3, 2026 for the QMSR. Using draft-era framing in your validation procedures signals to an auditor that your approach is out of date.
The fourth is treating CSA as a new regulation you must comply with. It is guidance on how to satisfy existing requirements, not a standalone rule. You comply with the QMSR and Part 11. CSA is the FDA's recommended, least-burdensome path to doing so for production and quality-system software.
The fifth is assuming CSA replaces GAMP 5. It does not. GAMP 5 categorizes software by how much you built versus inherited; CSA assures it by process risk. They answer different questions and work together, which is why the validation master plan should reference both the categorization approach and the risk-based assurance approach rather than choosing between them.
Where this leaves the engineer
CSA is not a new compliance vocabulary to memorize. It is regulatory permission to apply the judgment you already use. You do not test a limit switch as hard as a custom motion profile, and CSA says do not assure a boxed historian as hard as a custom batch-disposition routine. Determine the process risk of each software feature first. Match the assurance method to that risk: scripted and rigorous where a failure reaches the product, unscripted and efficient where it does not. Leverage the vendor evidence you are entitled to lean on. Record objective evidence proportionate to the risk, not screenshots proportionate to your anxiety. And keep the high-risk software, the custom code that can actually corrupt a record or release a bad batch, under the focused scrutiny it has always deserved and rarely got under the old uniform approach.
The scope of this still feeds your IQ, OQ, and PQ work and your change-control decisions, because the assurance you build is the evidence those activities rest on. CSA does not change what good validation is for. It changes where you spend the effort, and it finally puts the regulators on record that spending it evenly was never the goal.
---
Valiqa is an AI-powered validation lifecycle platform for regulated manufacturing. Learn more at valiqa.io
Frequently Asked Questions
Ready to automate your validation documentation?
Generate audit-ready IQ/OQ/PQ protocols in minutes, not weeks.
Get StartedWe use essential cookies for authentication and security. With your consent, we also use Microsoft Clarity, Google Analytics, and the LinkedIn Insight Tag on our marketing pages to understand how visitors navigate the site and to measure our advertising. Learn more.