How to Write a Part 11 Compliant CSV Protocol
Most Part 11 protocols fail in the same quiet way. Not at the audit, where the gaps finally surface, but on the day someone opened a generic CSV template, saw a heading that said "21 CFR Part 11 Compliance," and wrote a single test case under it: log in, confirm the audit trail recorded the login, pass. The system had electronic signatures, configurable password rules, a record-export function, and a sequencing lock on batch release, and none of it got tested, because the template treated Part 11 as one checkbox instead of the eleven distinct controls it actually is. The protocol looked complete. It proved almost nothing.
A computer system validation protocol does not make a system Part 11 compliant. Part 11 compliance is a set of controls the system and your procedures have to actually implement, and the protocol is the document where you demonstrate that the testable ones work and account for the ones that live in SOPs instead of software. This guide is for the engineer who has to write that document: how to scope Part 11 so you test what genuinely applies, how to separate the technical controls you verify from the procedural controls you reference, how to structure the protocol section by section, and how to build the requirements-to-test traceability that lets an auditor follow your logic from a regulation clause to a passed test step.
What Part 11 actually requires
21 CFR Part 11 is the FDA rule on electronic records and electronic signatures. It has been in effect since 1997, and it says that when you keep a required record electronically instead of on paper, or sign one electronically instead of by hand, the electronic version has to be as trustworthy and reliable as the paper one would have been. That is the whole purpose: electronic records that are attributable, protected, and tamper-evident, and electronic signatures that are genuinely bound to a real person and to the record they sign.
Two scoping facts decide how much of Part 11 your protocol actually has to address, and getting them right is what separates a focused protocol from a bloated or a negligent one.
The first is the predicate rule. Part 11 does not apply to every electronic record on the system. It applies to records you are required to keep by some other regulation, the predicate rule, that you have chosen to maintain electronically as the official record. For a medical device manufacturer, the predicate rule is now the Quality Management System Regulation, which took effect February 2, 2026 and incorporates ISO 13485:2016 by reference into 21 CFR Part 820. The QMSR did not change Part 11 itself. It changed which underlying records are required, and Part 11 rides on top of whatever those predicate records are. So the first question for any system is not "does this have records" but "does this hold electronic records that a predicate rule requires us to keep, and are they our official record." A configuration log that no regulation requires is not pulled into Part 11 scope just because it exists.
The second is the FDA's risk-based reading of the rule. The agency's 2003 guidance, "Part 11, Electronic Records; Electronic Signatures - Scope and Application," remains the current operative interpretation. In it the FDA stated that it would interpret the scope of Part 11 narrowly and would exercise enforcement discretion on certain requirements for certain systems, and it expects you to make Part 11 decisions, especially about validation depth, audit-trail scope, and record copies, on a documented, justified, risk basis tied to the record's importance and the predicate rule. (A separate 2023 guidance covers electronic systems in clinical investigations; it does not govern production and quality-system software, so do not pull it into a manufacturing protocol.) This is the same risk-based instinct that drives Computer Software Assurance, and the two fit together cleanly: CSA tells you how hard to test each function, Part 11 tells you which controls have to exist on the records that function touches.
One more distinction shapes the whole protocol: closed versus open systems. A closed system is one where access is controlled by the people responsible for the records, which covers nearly every on-premise SCADA, MES, LIMS, or eQMS behind your own authentication. Part 11 section 11.10 lists the controls for closed systems. An open system, where access is not controlled by the record owners, adds further measures under section 11.30, such as encryption and digital-signature standards. Most production systems are closed. State which one yours is in the protocol and justify it, because it determines which control set you test against.
The distinction that makes the protocol writable
Here is the move that turns Part 11 from an intimidating wall of clauses into a structured test plan: not every Part 11 requirement is a software feature you can test. Some are, and those belong in your test cases. The rest are procedural and administrative controls that live in SOPs, training records, and a signed certification, and those belong in the protocol as documented references, not as executed steps. A protocol that tries to "test" a training program is confused. A protocol that forgets to verify the audit trail is dangerous. Sorting every requirement into the right bucket is most of the work.
The technical controls, the ones your protocol verifies with real test steps, are the parts of Part 11 the system enforces in software:
- Access control and authority checks (11.10(d) and (g)). These are two linked controls: 11.10(d) limits system access to authorized individuals, while 11.10(g) enforces authority checks at the transaction level so that an authenticated user is still allowed only the operations their role permits, such as signing, altering, or deleting a record. Tested by exercising each role against permitted and forbidden actions at both the system-entry level and the individual-operation level.
- Device checks (11.10(h)), where applicable. The system verifies the validity of the source of a data input or operational instruction, as appropriate, for example confirming that a reading came from the qualified instrument it claims to come from. Many systems can justifiably scope this out, but the decision has to be a documented one. Tested where implemented, or excluded with a rationale in the applicability assessment rather than silently omitted.
- Audit trail (11.10(e)). A secure, computer-generated, time-stamped trail records who did what and when for create, modify, and delete actions, and changes do not obscure prior values. Tested by making changes and confirming the trail captured them, attributed them, time-stamped them, and preserved the old value.
- Accurate copies (11.10(b)). The system produces accurate and complete copies of records in both human-readable and electronic form for the agency. Tested by exporting and reconciling against the source.
- Record protection and retrieval (11.10(c)). Records remain accurately retrievable across the retention period. Tested in part through backup and restore verification.
- Operational and sequencing checks (11.10(f)). The system enforces permitted step sequencing where it matters, such as not allowing release before approval. Tested by attempting to break the sequence and confirming the system blocks it.
- Signature manifestation (11.50). A signed record shows the signer's printed name, the date and time, and the meaning of the signature (review, approval, responsibility). Tested by signing and inspecting the rendered record and its export.
- Signature-to-record linking (11.70). Signatures are bound to their records so they cannot be cut, copied, or transferred to falsify. Tested by confirming the binding holds through export and that signatures cannot be lifted onto another record.
- Electronic signature components (11.200). Non-biometric signatures use at least two distinct identification components, such as a user ID and a password. Tested by confirming both are required and a single component is rejected.
- Identification code and password controls (11.300, testable parts). Uniqueness of each ID and password combination (11.300(a)), password aging and revision (11.300(b)), and the transaction safeguards that prevent and detect unauthorized use, such as lockout and alerting on repeated failed attempts (11.300(d)). Tested through the configurable account and password policy.
The procedural and administrative controls, the ones your protocol references rather than executes, are the parts Part 11 requires but software cannot prove on its own:
- Validation itself (11.10(a)). The requirement to validate the system for accuracy, reliability, and consistent intended performance is satisfied by the existence and execution of this protocol. You do not test it; you are it.
- Personnel qualification (11.10(i)). The people who develop, maintain, and use the system have the education, training, and experience for their tasks. Referenced through training records, not a test case.
- Accountability policies (11.10(j)). Written policies hold individuals responsible for actions taken under their electronic signatures. Referenced through the relevant SOP.
- Systems documentation controls (11.10(k)). Distribution, access, and revision control over system documentation. Referenced through document-control procedures.
- Identity verification and the FDA certification (11.100). Each signature is unique to one person whose identity was verified before the signature was issued, and the organization has certified to the FDA in writing that its electronic signatures are the legally binding equivalent of handwritten ones. Referenced through your identity-proofing procedure and the certification letter on file, not through a software test.
- Credential loss management and device testing (11.300(c) and (e)). The procedures for deauthorizing lost, stolen, or compromised tokens and cards and issuing controlled replacements (11.300(c)), and the periodic testing of any physical tokens or devices that carry credentials (11.300(e)), where your system uses them. Referenced through the account-management and device SOPs, while the software-enforced parts of 11.300 are tested above.
The protocol's job is to verify every control in the first list and explicitly account for every control in the second, so that nothing in Part 11 is left without either a test result or a documented procedural home. The traceability matrix is where you prove you did both.

Set the rigor before you write a line
Two decisions made before you draft the protocol determine how heavy each test case should be, and skipping them is how protocols end up either thin where it counts or bloated where it does not.
First, categorize the software. The GAMP 5 category tells you how much of the system you built versus inherited from a competent supplier. A configured commercial eQMS used as shipped is not validated the way a custom batch-release routine is. The category sets your baseline effort and tells you how much vendor evidence you are entitled to lean on instead of re-proving platform behavior the supplier already tested.
Second, assess process risk per function the way CSA directs. A function whose failure could release a bad batch or corrupt a quality record earns rigorous scripted testing with predefined steps and expected results. A low-risk function earns lighter, justified methods. The Part 11 controls do not all carry the same weight either: the audit trail and signature binding on a batch-disposition record deserve more scrutiny than the same controls on a low-consequence log. Let GAMP 5 and CSA size each test case, then write the protocol against that sizing rather than applying uniform depth to everything. This is also why the protocol's approach belongs in your validation master plan, which is where the categorization and risk-based assurance strategy are supposed to be declared.
The protocol, section by section
A Part 11 CSV protocol is a structured validation document, so it carries the usual scaffolding plus a Part 11 backbone. The sections that matter:
Purpose and scope. State the system, its version, the boundaries of what you are validating, and explicitly whether it is a closed or open system. Name the predicate rule whose records the system holds. This is where you record the Part 11 applicability decision so the rest of the protocol has a foundation.
System description. What the system does, its components, interfaces, where its electronic records live, and which records are predicate-rule records under Part 11 scope. An auditor reads this to understand what should have been tested.
Roles and responsibilities. Who authored, reviewed, executed, and approved, and who owns the system and its quality oversight. Part 11 records are signed off by people; name them.
References and standards. Part 11 itself, the predicate rule (the QMSR and the applicable ISO 13485:2016 clause for the records in question), the FDA scope-and-application guidance, GAMP 5, CSA, and your governing SOPs. This is also where the procedural controls get their home: the training procedure, the account-management SOP, the document-control procedure, and the electronic-signature certification letter all get cited here so the protocol can point to them instead of testing them.
Part 11 applicability assessment. A short, explicit analysis: which electronic records on this system are predicate-rule records, whether electronic signatures are used and for what, closed versus open, and the risk-based justification for the depth of testing you are about to apply. This section is what an auditor reads first to judge whether your scope was honest.
Requirements traceability matrix. The core of the document. Every applicable Part 11 control mapped to either a test case that verifies it or a procedural control that satisfies it. More on its structure below.
Test approach (IQ, OQ, PQ). Installation qualification confirms the system is installed and configured correctly, including the security and audit-trail configuration. Operational qualification is where most Part 11 technical controls are exercised: access, audit trail, signatures, sequencing, copies. Performance qualification confirms the system performs against the controls under real or representative use. The same IQ, OQ, and PQ structure you use for equipment applies to software, with the Part 11 controls slotted predominantly into OQ.
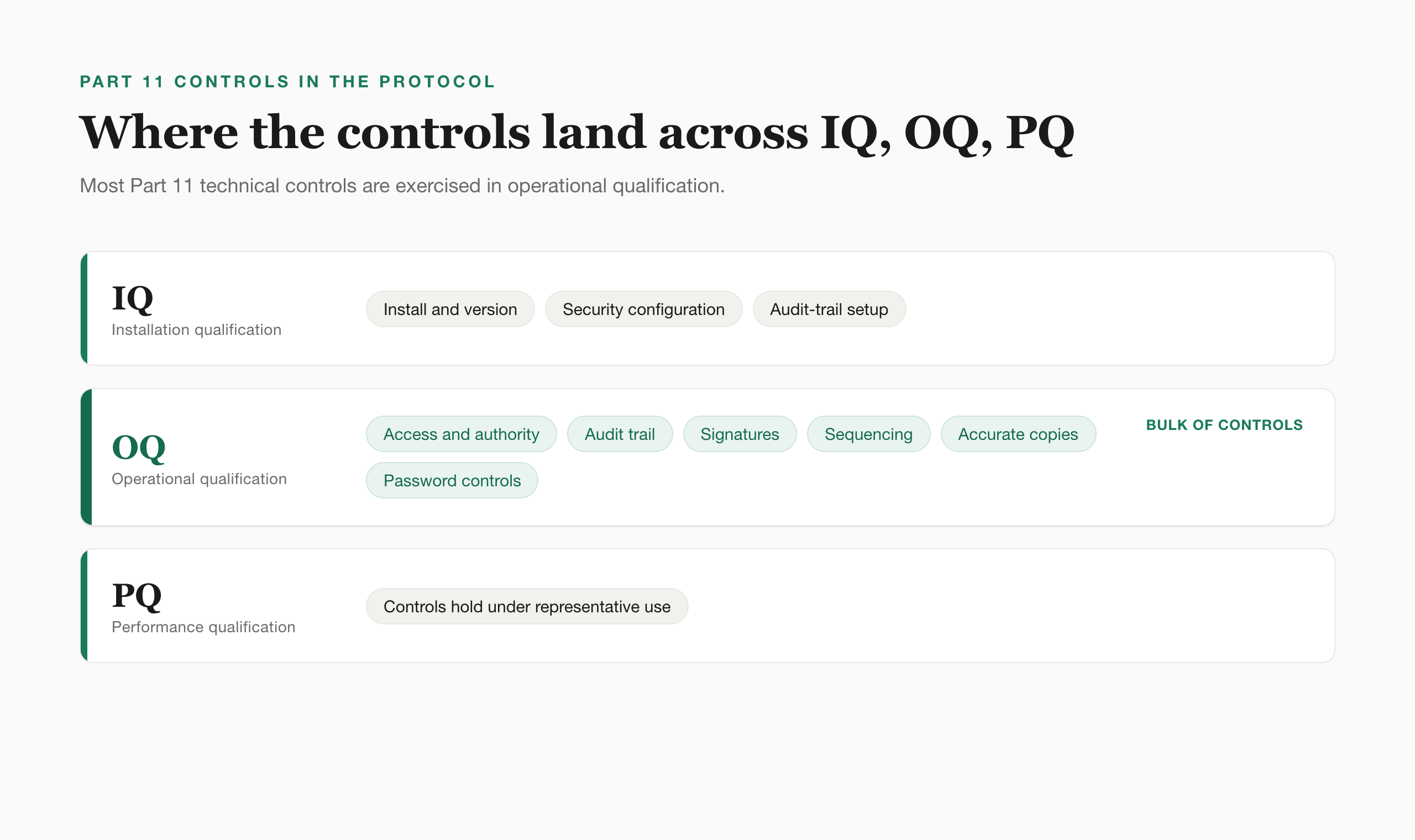
Test cases. Each with a unique ID, the Part 11 clause it traces to, preconditions, steps, expected results, and a place to record actual results, pass or fail, evidence reference, executor, and date. Write acceptance criteria that are specific and pre-stated, not "audit trail works" but "the audit trail records the user ID, date, time, old value, and new value for a modification, and the prior value remains visible."
Deviation handling. How a failed step is documented, assessed, and resolved before the protocol can be approved. A failed Part 11 control test, such as an audit-trail gap, feeds directly into the 11.10(a) validation conclusion and usually triggers a risk assessment before the system can be released. Auditors expect deviations to exist and to be handled, not hidden.
Summary and approval. The conclusion that the system is validated and fit for its intended use under Part 11, with the signatures that close it out.
The traceability matrix is the deliverable
If an auditor remembers one artifact from your protocol, it will be the matrix that lets them trace a Part 11 clause to the evidence that it was satisfied. Build it as a table with one row per control, each row stating the clause, what it requires, whether it is verified by test or satisfied by procedure, and the pointer to the test case ID or the SOP. A workable shape:
| Part 11 clause | Requirement | How satisfied | Trace |
|---|---|---|---|
| 11.10(a) | System validated for intended use | This protocol | Whole protocol |
| 11.10(b) | Accurate, complete copies for the agency | Test | OQ-PART11-01 |
| 11.10(c) | Records protected and retrievable through retention | Test | OQ-PART11-02 (backup/restore) |
| 11.10(d), (g) | Access limited to authorized individuals and roles | Test | OQ-PART11-03 |
| 11.10(e) | Secure, time-stamped audit trail; prior values preserved | Test | OQ-PART11-04 |
| 11.10(f) | Operational sequencing checks enforced | Test | OQ-PART11-05 |
| 11.10(h) | Device/terminal checks on source of input, as appropriate | Test, or scope out with rationale | OQ-PART11-10 or applicability assessment |
| 11.10(i) | Personnel qualified by education, training, experience | Procedure | Training SOP ref |
| 11.10(j) | Accountability policy for signed actions | Procedure | Accountability SOP ref |
| 11.10(k) | Systems documentation controlled | Procedure | Doc-control SOP ref |
| 11.50 | Signature shows name, date/time, meaning | Test | OQ-PART11-06 |
| 11.70 | Signatures linked to records, not transferable | Test | OQ-PART11-07 |
| 11.100 | Signatures unique, identity verified, FDA certification filed | Procedure | Identity SOP + certification letter ref |
| 11.200 | Two distinct signature components enforced | Test | OQ-PART11-08 |
| 11.300(a), (b), (d) | ID/password uniqueness, aging, transaction safeguards against unauthorized use | Test | OQ-PART11-09 |
| 11.300(c), (e) | Loss management for lost/stolen credentials; device testing where tokens are used | Procedure | Account-management and device SOP ref |
Two things make this matrix audit-proof rather than decorative. Every "Test" row must point to a real test case whose acceptance criterion actually exercises the control, not a token login. And every "Procedure" row must point to a procedure that genuinely exists and is in force, because an auditor will sometimes pull the SOP you cited. The matrix is a set of promises; both columns have to be kept.
A worked example: validating an eQMS module
Take a configured electronic quality management system used to author, review, and electronically approve controlled documents and CAPA records. The approved documents and CAPA records are required by the QMSR, kept electronically as the official record, and signed electronically. That puts the system squarely in Part 11 scope as a closed system. By GAMP 5 it is largely configured commercial software, so you lean on the vendor's platform validation for the underlying engine and focus your testing on your configuration, your workflows, and the Part 11 controls as you have set them up.
Start with the applicability assessment: the controlled-document and CAPA records are predicate-rule records, electronic signatures are used for review and approval, the system is closed, and the risk is meaningful because an unapproved or altered procedure reaching the floor is a real quality event. That justifies rigorous scripted testing of the Part 11 controls rather than a light touch.
Then the test cases follow the matrix. For the audit trail, you modify a document in draft, change an approval, and delete a CAPA action, then confirm the trail captured each action with the user, the date and time, and the prior value, and that nothing overwrote history (11.10(e)). For signatures, you approve a document and inspect the manifestation for the signer's printed name, the date and time, and the meaning shown as "Approved," then export the record and confirm the signature stays bound and legible (11.50, 11.70). For the signature components, you confirm that approval requires both the user ID and the password and that supplying only one is rejected (11.200). For access, you exercise an author, a reviewer, and an approver against actions each should and should not be able to perform, including confirming an author cannot approve their own document where your workflow forbids it (11.10(d), (g), and the sequencing check at 11.10(f)). For copies, you generate the human-readable and electronic exports an inspector would request and reconcile them against the source (11.10(b)). Backup and restore verifies retrievability across retention (11.10(c)).
The procedural rows do not get test cases. The training requirement is met by your training records and cited (11.10(i)). The uniqueness and identity-verification requirement is met by your account-provisioning SOP and the electronic-signature certification letter your organization filed with the FDA, both referenced in the protocol (11.100). What the software enforces about passwords, you test; what the people and procedures enforce about credentials, you cite. When the protocol is executed, every Part 11 control has either a passed test step or a named procedure behind it, and the matrix shows an auditor exactly which is which.
Common mistakes that get protocols flagged
The first and most common is treating Part 11 as a single test case. "Verify Part 11 compliance: pass" verifies nothing. Part 11 is a set of distinct controls, and each applicable one needs its own traced verification or its own documented procedure.
The second is testing controls the system does not actually have in scope, or skipping ones it does. A protocol that writes elaborate electronic-signature test cases for a system that uses no electronic signatures is wasting effort; one that skips the audit-trail test on a system full of predicate-rule records is exposed exactly where it matters. The applicability assessment is what keeps you honest in both directions.
The third is confusing the procedural controls for testable ones, or worse, forgetting them. You cannot software-test a training program or an identity-verification process, and pretending to is a red flag. But leaving 11.100 or 11.10(i) entirely unaddressed because they are not testable is just as wrong. They belong in the matrix as procedural rows with real references.
The fourth is vague acceptance criteria. "Audit trail functions correctly" is not a criterion an auditor can confirm you met. State exactly what the trail must capture and what must remain visible, the same discipline that keeps any acceptance criterion from getting flagged.
The fifth is writing the protocol as if validation ends at approval. When the eQMS gets a configuration change or a platform upgrade, the Part 11 controls have to be re-assured proportionate to the change, the same judgment that decides whether a software update triggers revalidation. A protocol that never anticipates change leaves the next engineer guessing.
Where this leaves the engineer
A Part 11 compliant CSV protocol is not a special document type with secret rules. It is an ordinary validation protocol that has been disciplined by one clear idea: Part 11 is a list of specific controls, each control is either something the software enforces or something your procedures enforce, and the protocol's job is to prove the first kind works and account for the second kind in writing. Scope it with the predicate rule and the risk-based reading so you test what genuinely applies. Sort every control into technical or procedural. Let GAMP 5 and CSA size the rigor. Build the traceability matrix so an auditor can walk from any clause to its evidence. Write acceptance criteria precise enough that pass and fail are not matters of opinion. Do that, and the protocol stops being a checkbox someone hopes covers the regulation, and becomes what it was always supposed to be: the documented, defensible proof that your electronic records and signatures can be trusted.
---
Valiqa is an AI-powered validation lifecycle platform for regulated manufacturing. Learn more at valiqa.io
For device manufacturers grounding protocols in the standard itself, see how to generate ISO 13485 validation protocols.
Frequently Asked Questions
Ready to automate your validation documentation?
Generate audit-ready IQ/OQ/PQ protocols in minutes, not weeks.
Get StartedWe use essential cookies for authentication and security. With your consent, we also use Microsoft Clarity, Google Analytics, and the LinkedIn Insight Tag on our marketing pages to understand how visitors navigate the site and to measure our advertising. Learn more.