What Is a Validation Master Plan and Who Owns It?

If your last audit ended with the inspector asking for the Validation Master Plan and your quality director quietly pulling up a folder no one had touched in two years, you already know the problem. The VMP is often one of the first artifacts inspectors use to orient on a validation program, and it is the artifact most teams have stopped maintaining. It is either too thin to defend a real validation program or too thick to be a working document. Neither version helps when the inspector wants to see how your team actually decides what to qualify and what evidence supports the qualification.
This guide walks through what a VMP actually is, who owns it in a healthy organization, the sections a defensible VMP contains, where teams get this wrong, and how the VMP fits under current US and EU regulatory frameworks.
What a Validation Master Plan actually is
A Validation Master Plan is a single controlled document that describes how an organization runs its validation program: what gets validated, why, against what standards, by whom, on what cadence, and how changes and deviations are handled. It is strategy and policy, not execution. The individual IQ, OQ, PQ, PPQ, CSV, and PV protocols are governed by the VMP; the VMP itself never executes a test step.
A useful working definition: the VMP is the document that lets a new validation engineer joining the company understand the validation philosophy of the organization without asking anyone. It captures decisions that would otherwise live in people's heads. EU GMP Annex 15 §1.4 puts the same point more formally: the key elements of the qualification and validation programme should be clearly defined and documented in a VMP or equivalent document.
Three properties separate a defensible VMP from a doorstop. The VMP is risk-based, meaning the scope of validation effort is justified by the risks the validated systems present. The VMP is living, meaning it changes when the organization, regulations, or equipment portfolio change. And the VMP is integrated with the QMS, meaning it references and is referenced by the procedures that actually run the day-to-day work, rather than duplicating their content.
A VMP that fails any of those three properties is a compliance artifact, not a working document. Compliance artifacts get found during audits and never get used in between.
Who actually owns the Validation Master Plan
Ownership of the VMP is one of the most consistently muddled questions in regulated manufacturing. The defensible answer depends on the size of the organization, but it follows a clear pattern.
In a small to mid-size organization, the VMP is owned by Quality, drafted by Validation Engineering, and signed by Operations and Quality Assurance leadership. Quality owns it because the VMP describes how the organization assures product quality through validated systems. Validation Engineering drafts it because they are the team that understands what each system actually needs. Operations signs because operations runs the equipment day to day. Quality Assurance leadership signs because they are accountable to regulators.
In a larger organization with a dedicated validation function, the VMP is owned by the Validation Director or equivalent, with Quality reviewing and signing. The function-level ownership matters because at scale, the VMP needs an organizational home with continuity and authority to enforce its contents.
Cross-functional review of the VMP is normal and healthy in mature organizations; what is not healthy is committee-only ownership with no single accountable owner. Without a clear owner, the VMP drifts because every update requires multiple sign-offs from people whose primary jobs are elsewhere. Committees can review and approve VMPs effectively, but the maintenance has to land on one accountable function.
A second dysfunction signal is the consultant-drafted VMP whose ownership was never transitioned internally after the engagement ended. Consultant-drafted VMPs are common and not inherently bad, but ownership has to land with an internal function from day one, with the consultant document treated as a starting point rather than the final artifact.
What a defensible VMP contains
A defensible VMP has a consistent shape regardless of company size. The order and depth varies; the categories do not. We have seen ten sections show up in nearly every audit-ready VMP we have read.
1. Scope and applicability
What the VMP covers, what it explicitly does not cover, and which sites, products, processes, and computerized systems are governed by this document. Out-of-scope items get a one-sentence rationale and a pointer to the document that does cover them. A VMP that does not state its scope clearly is a VMP that an auditor will pick at first.
2. Regulatory framework and validation policy
The predicate regulations, consensus standards, and industry guidance documents this organization's validation program is built against, plus a short statement of the organization's qualification and validation policy. Annex 15 §1.5 lists policy as expected VMP content; teams that skip it are skipping something inspectors look for.
The framework set depends on industry and geography.
For a US medical device manufacturer, the anchors are 21 CFR Part 820 under the FDA Quality Management System Regulation, effective February 2, 2026, which incorporates ISO 13485:2016 by reference. ISO 13485:2016 §7.5.6 is the relevant process-validation clause, though the VMP itself is an organizational implementation choice rather than an explicit regulatory requirement. 21 CFR Part 11 applies as an electronic-records and electronic-signatures overlay where required records or signatures are kept electronically. For EU market access on medical devices, EU Regulation 2017/745 (MDR), ISO 13485, and MDSAP for harmonized inspections of medical device QMS are the applicable framework. EudraLex Volume 4 (including Annex 15 and Annex 11) governs medicinal products and is not the medical-device anchor.
For a US pharmaceutical manufacturer, the anchors are 21 CFR Part 210/211 plus 21 CFR Part 11 where electronic records and signatures are involved. The FDA process validation guidance describes the Stage 1, Stage 2, and Stage 3 lifecycle that a pharmaceutical VMP organizes around. For EU pharmaceutical operations, EudraLex Volume 4 applies, including EU GMP Annex 15 (qualification and validation) and Annex 11 (computerized systems).
Industry guidance worth citing across both worlds includes GAMP 5 and the FDA's computer software assurance approach for computerized systems, and ICH Q9 for risk management. Where medical device process validation is in scope, GHTF SG3/N99-10 is sometimes referenced as legacy guidance; IMDRF has noted that the older GHTF documents contain outdated principles, so use them carefully.
The framework section is not a list of every regulation that exists. It is the specific set that govern this organization's validation activities, with a sentence explaining why each applies, plus the policy statement that ties the organization's approach back to those references.
3. Validation strategy, risk approach, and acceptance criteria principles
How the organization decides what to qualify, what depth of qualification, and what evidence is sufficient. This section is where ICH Q9 risk principles live: severity-occurrence-detection assessment, risk-based scoping of test plans, the relationship between equipment criticality and validation effort. Annex 15 §1.5 also expects the VMP to describe the principles for developing acceptance criteria, so this section captures how the organization derives criteria from URS, design inputs, and specifications rather than restating per-protocol thresholds. A VMP without an explicit risk approach and an acceptance-criteria philosophy is a VMP that cannot defend why it tested one piece of equipment heavily and another lightly, or how it set the bar for "pass." We covered the practical side of acceptance criteria in our post on acceptance criteria that won't get flagged in an audit.
4. Inventory, validation status matrix, and plan hierarchy
A list of every qualified asset under the VMP's scope, with criticality classification, current qualification status, and a pointer to the active protocols and reports. This is often a table or a reference to a controlled inventory maintained elsewhere in the QMS.
For larger or multi-site organizations, this section also describes the plan hierarchy: the VMP at the top, with subordinate site-level or project-level Validation Plans that govern specific scopes underneath it. The status matrix shows, asset by asset, whether qualification is current, scheduled, in progress, or overdue, with the responsible owner and the next periodic review date. Either the matrix lives in the VMP or the VMP references the controlled document where it does live; what is not acceptable is no matrix at all.
5. Roles and responsibilities
A responsibility matrix showing who does what across Quality, Validation Engineering, Operations, Regulatory Affairs, and where applicable IT and Facilities. The matrix typically uses RACI or a similar framework. The point is that during an audit, the inspector should be able to read this section and know exactly who they need to talk to about any specific aspect of the validation program.
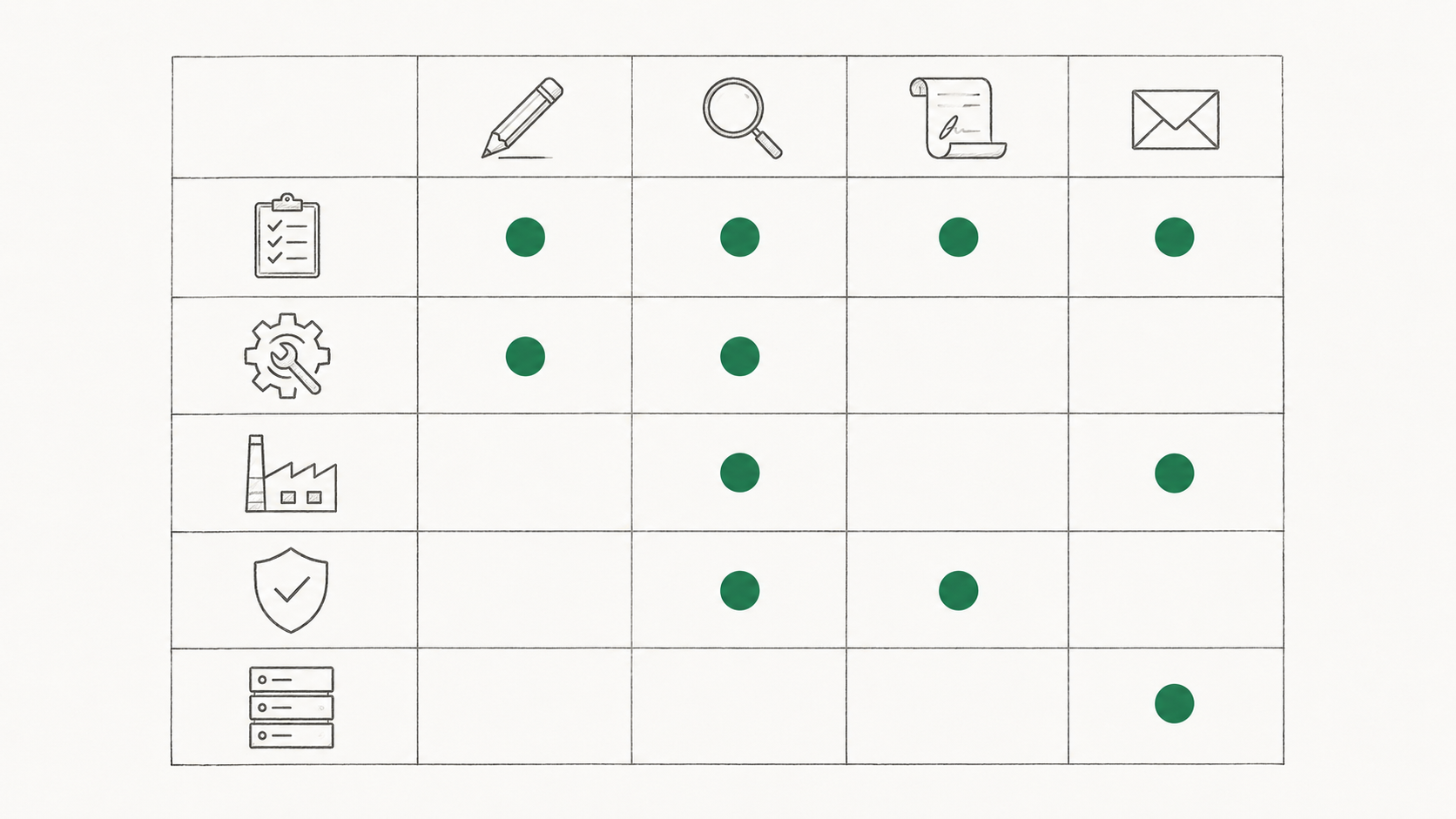
6. Document types and templates
The list of protocol and report types the organization uses (IQ, OQ, PQ, DQ, PPQ, CSV, CSA, PV, TQ, plus VMP and PFMEA as documents under the VMP), with their controlled template references and a brief description of when each applies. We covered the substance of these document types in our pillar post on IQ vs OQ vs PQ and what actually goes in each one. For the process side specifically, the PPQ protocol guide walks through a process performance qualification step by step.
7. Change control and revalidation approach
How changes to validated systems are evaluated, scoped, and re-validated. This section references the change control SOP and describes the risk-based criteria for deciding scope. We covered the revalidation scope decision in detail in our post on revalidation: when do you actually need to redo IQ/OQ/PQ.
8. Deviation, nonconformance, and CAPA approach
How validation-related deviations and findings are captured, investigated, and resolved. This section typically references the broader QMS deviation and CAPA procedures rather than duplicating them.
9. Periodic review and ongoing oversight
The cadence and scope of periodic review for qualified assets. For pharmaceutical manufacturers, the section also describes the organization's approach to FDA Stage 3 Continued Process Verification, which is a pharma-specific term from the FDA process validation guidance. Medical device contexts typically describe ongoing monitoring or periodic review without the CPV label. Either way, this is the section that signals whether the organization has moved past calendar-based annual revalidation into a modern lifecycle approach.
10. Training and document control
How validation engineers are qualified to perform validation work, how SOPs and templates are controlled, and how training records are maintained. Auditors connect training records to executed protocols; a VMP that does not address training assumes that connection is documented elsewhere.
Where teams get the VMP wrong
A few failure modes account for most VMP problems we see in real organizations.
The first is the template-copy VMP. A team downloads a template from a regulatory training course or copies a VMP from a prior employer, fills in the blanks, and never updates it. The document reads plausibly to anyone unfamiliar with the company; it reads transparently to an auditor who notices that the regulatory references are dated, the equipment inventory does not match the floor, and the risk approach references a methodology nobody on the team has used. The fix is not a better template. The fix is treating the VMP as a strategy document that the team actually built rather than inherited.
The second failure mode is the VMP without a risk approach. The document describes what gets validated but not why. Every qualification is full IQ/OQ/PQ regardless of equipment criticality. Auditors do not credit this approach because it does not match how regulators expect risk to be applied. ICH Q9, GAMP 5, and FDA's process validation guidance all assume the organization scopes effort to risk. A VMP without a risk approach signals that the team has not internalized the framework.
The third failure mode is the dead VMP. The document exists, is technically up to date, but nobody on the team consults it during day-to-day work. Validation engineers make scope decisions by feel because the VMP does not actually answer their questions. Quality reviews protocols against personal experience rather than the VMP's stated approach. The document is compliant; the program is not aligned. The signal is asking three engineers on the team to describe the validation strategy and getting three different answers.
The fourth failure mode is the over-engineered VMP. The opposite of the dead VMP. The document tries to be a single source of truth for every validation activity in the organization, duplicates content from SOPs, and grows to several hundred pages. Maintaining it becomes a quarterly project that nobody owns. The fix is the discipline to reference the QMS rather than duplicate it, keeping the VMP focused on strategy and policy.
VMP under QMSR, EU frameworks, and ICH
The VMP sits at the intersection of three regulatory layers, and a defensible document acknowledges all three explicitly. Which set of citations applies depends on industry and geography.
The predicate regulation layer is the strongest anchor. For US medical device manufacturers, Part 820 under the FDA Quality Management System Regulation (effective February 2, 2026) incorporates ISO 13485:2016 by reference. The VMP is not named explicitly in QMSR text and is best understood as an organizational implementation choice that satisfies the broader QMS expectations, not a separately required document. The pre-QMSR QS Regulation expressed the related process-validation requirement at 21 CFR 820.75(c), which teams with documentation predating the transition will still see in their controlled copies. For US pharmaceutical manufacturers, 21 CFR Part 210/211 carries equivalent process-validation expectations; the FDA does not explicitly require a document titled VMP, but most drug manufacturers use one to organize and document their validation approach.
The consensus standards layer is mostly ISO 13485 and ISO 14971 for medical device manufacturers. ISO 13485 sets the QMS framework; ISO 13485:2016 §7.5.6 is the relevant process-validation clause. ISO 14971 governs the risk management process that feeds the VMP's risk approach. Pharma manufacturers reference the relevant ISO and ICH standards depending on product type.
The industry guidance layer is where the VMP gains practical substance, and this is where the layer applies differently across industries. EU GMP Annex 15 (within EudraLex Volume 4) is the most explicit single source on validation master planning, naming the VMP directly and describing its expected contents in §1.4 and §1.5. Annex 15 governs medicinal products, so it applies to pharmaceutical and biotech operations in the EU; it is not the right anchor for medical devices. EU Annex 11 covers computerized systems for medicinal products. For EU medical device market access, the framework is instead EU MDR 2017/745, ISO 13485 implementation, and MDSAP for harmonized QMS inspections. GAMP 5 fills in the practical detail for computer system validation strategy across both worlds. ICH Q9 provides the risk-management framework. FDA's process validation guidance describes the Stage 1, Stage 2, and Stage 3 lifecycle that pharmaceutical VMPs organize around.
A VMP that cites the predicate regulation, the relevant consensus standards, and the applicable industry guidance, in the right hierarchy and matched to the actual industry, is a VMP that reads as competent. A VMP that misapplies Annex 15 to medical devices or treats Part 11 as the primary CSV anchor instead of an electronic-records overlay is a VMP that signals the team has not worked through the framework systematically.
The VMP as a living document
A defensible VMP is updated on triggers, not on a calendar. The triggers are familiar to anyone who has worked through a revalidation scope decision.
A new piece of qualified equipment or a new validated process enters the program. The VMP equipment inventory is updated and any new validation strategy considerations are addressed.
A regulatory framework changes. The QMSR transition in February 2026 is the recent example for medical device manufacturers; teams that updated their VMP regulatory framework section during that transition demonstrated current alignment. Teams that did not are signaling to auditors that the VMP has not been touched.
A process or system change with VMP-level implications occurs. Not every change triggers a VMP update, but changes that affect validation strategy or the scope of the validation program do. New product lines, new sites, major reorganizations of the validation function, or changes to risk approach all qualify.
A periodic review finds that the VMP no longer matches how the organization actually operates. This is the most common legitimate trigger and the most commonly skipped. Periodic VMP review is an opportunity to surface drift between the document and the practice. The output should be either confirmation that they match or a documented set of updates.
The mechanics are mundane: change control on the VMP itself, document control approval, version increment, training where the changes affect how the team operates. The discipline is what separates the living document from the doorstop.
A worked example
Consider a mid-size pharmaceutical manufacturer with one site, two solid-dose production lines, twenty qualified pieces of equipment, and one validated electronic batch-record system. The product is sold into both US and EU markets. Their VMP table of contents looks like this:
Section 1: Scope. Names the site, the two production lines, the equipment inventory by category, and the computerized batch record system. Excludes the analytical method validation program (which is governed by a separate documented procedure under the QC organization) and the development laboratory (covered by a separate development qualification policy).
Section 2: Regulatory framework and validation policy. Cites 21 CFR Part 210/211 as the US predicate regulation, EudraLex Volume 4 Part I plus Annex 15 and Annex 11 for the EU market, 21 CFR Part 11 as the electronic-records overlay for the batch record system, ICH Q9 for risk principles, ICH Q10 for the pharmaceutical quality system context, and GAMP 5 as the industry guidance for the computerized system. The FDA process validation guidance is referenced for the Stage 1, Stage 2, and Stage 3 lifecycle. A short qualification and validation policy statement closes the section, expressing the organization's commitment to a risk-based lifecycle approach.
Section 3: Validation strategy, risk approach, and acceptance criteria principles. Describes the risk-based methodology aligned to ICH Q9, the company's process for documented criticality classification, and the philosophy for deriving acceptance criteria from URS and design inputs. References the risk management procedure for the operational detail.
Section 4: Inventory, validation status matrix, and plan hierarchy. A table with twenty equipment rows and one system row. Each row lists the criticality, the current qualification status, the active protocol or report numbers, the date of last requalification, and the next periodic review date. The VMP is the top-level document; below it sit project-level validation plans for any active new-product introduction or capital project.
Section 5: Roles and responsibilities. A RACI-style matrix covering Quality, Validation Engineering, Operations, Maintenance, IT (for the computerized system), and Regulatory Affairs.
Section 6: Document types and templates. Lists IQ, OQ, PQ, PPQ, CSV, PV, and PFMEA, with the controlled template numbers and a one-sentence description of when each applies.
Section 7: Change control approach. References the company's change control SOP. Describes the risk-based scoping criteria for requalification decisions (the substance of which we covered in our revalidation post).
Section 8: Deviation and CAPA approach. References the company's QMS deviation and CAPA procedures.
Section 9: Periodic review and continued process verification. Describes the annual periodic review process for each qualified asset and the company's Stage 3 Continued Process Verification program for the validated process.
Section 10: Training and document control. References the training SOP and the document control SOP.
The whole document runs in the dozens of pages, illustratively, with the exact length depending on whether appendices and the validation status matrix sit inside the VMP or in referenced controlled documents. It cites and references the QMS rather than duplicating it. The section list aligns with Annex 15 §1.5 expectations and would survive an inspection because every section answers a question an inspector might ask, and every section is anchored either in the regulatory framework or in another controlled document.
If the same organization were a medical device manufacturer rather than pharma, Section 2 would substitute Part 820 under the FDA QMSR (with ISO 13485:2016 incorporated by reference) and EU MDR 2017/745 plus MDSAP for the EU market, in place of Part 210/211 and Annex 15/11. The rest of the section structure stays the same.
Further reading
- Validation Master Plan template (free download)
- IQ vs OQ vs PQ and what actually goes in each one
- Revalidation: when do you actually need to redo IQ/OQ/PQ
- How to write acceptance criteria that won't get flagged in an audit
- What auditors look for in validation documentation
- How to choose validation software for equipment qualification
- Self-scoring tool: which category of validation software fits your team
The VMP is not the most exciting validation document, and that is the point. It is the strategy document that lets the executed protocols make sense to anyone who reads them in sequence. A working VMP makes new validation engineers productive faster, helps inspections orient cleanly on the program, and makes scope decisions defensible. A dead VMP does none of those things and quietly costs the organization in every audit and every onboarding.
Frequently Asked Questions
Ready to automate your validation documentation?
Generate audit-ready IQ/OQ/PQ protocols in minutes, not weeks.
Get StartedWe use essential cookies for authentication and security. With your consent, we also use Microsoft Clarity on our marketing pages to understand how visitors navigate the site. Learn more.