Cleaning Validation: Worst Case, Carryover Limits, and Sampling That Holds Up
Shared equipment is where contamination risk concentrates. The mixer that ran a high-potency product on Monday runs a different product on Tuesday, and the only thing standing between those two batches is a cleaning procedure and the evidence that it works. Cleaning validation is that evidence. Done well, it is a defensible chain from a health-based limit to a sampling result. Done poorly, it is a binder of swab numbers with no rationale for why those locations, that limit, or that product were chosen, and inspectors know exactly where to push.
This guide is for the engineer or quality lead who owns a cleaning validation program and needs it to survive scrutiny. It covers how to select the worst case, how to set carryover limits using health-based exposure data instead of legacy rules of thumb, how to choose and defend swab versus rinse sampling, how recovery studies underpin every result, and how to justify dirty and clean hold times. The regulatory frame is the current one, not the one from a decade ago.
What cleaning validation actually demonstrates
Cleaning validation is documented evidence that a cleaning procedure consistently reduces product residue, cleaning agent residue, and microbial load on shared product-contact surfaces to a level that is safe for the next product. The three residue classes matter equally: the previous product's active ingredient, the detergent or solvent used to clean, and bioburden or endotoxin on surfaces that sit wet between runs.
The word that carries the weight is consistently. A single clean that passes proves nothing about the procedure. Cleaning validation, like process validation, rests on demonstrating reproducibility, which is why the standard expectation is a defined number of consecutive successful cleaning runs executed against a written protocol with predetermined acceptance criteria. The runs have to be consecutive because a procedure that passes on three of five attempts is not a validated procedure, it is an unreliable one with a favorable sample.
Cleaning validation lives inside the broader validation lifecycle. It draws its scope from risk assessment, it references the same change control and revalidation triggers as equipment qualification, and its limits flow from toxicology. If you are building the surrounding program, the Validation Master Plan is where the cleaning validation strategy, the grouping approach, and the revalidation triggers get documented.
The regulatory frame, stated correctly
Three references govern most cleaning validation programs, and using the current versions matters because the acceptance-limit basis changed materially in the last decade.
EU GMP Annex 15, Section 10, sets the core expectations for cleaning validation in medicinal products: validate the actual cleaning procedure, base limits on toxicological evaluation, justify sampling methods with recovery data, and address both dirty and clean hold times. Annex 15 is the most specific single source and is worth reading in full if cleaning validation is your responsibility.
Health-based exposure limits are the current basis for carryover limits. The EMA guideline on setting health-based exposure limits (EMA/CHMP/CVMP/SWP/169430/2012, effective 2015) established the Permitted Daily Exposure, or PDE, sometimes expressed as an Acceptable Daily Exposure or ADE, as the toxicologically derived amount of a substance a person can be exposed to daily without appreciable risk. This replaced the older practice of relying solely on the 1/1000th-of-a-dose criterion and the 10 parts-per-million criterion as the sole basis for limits. Those legacy criteria still appear in programs and can serve as additional checks, but a modern limit is anchored to a PDE derived by a qualified toxicologist. The ISPE Baseline Guide on Risk-Based Manufacture of Pharmaceutical Products, commonly called Risk-MaPP, is the practical companion for applying health-based limits to shared facilities.
For US drug manufacturers, the FDA expectation flows from the process validation framework and the long-standing agency guidance on inspecting cleaning process validation, which emphasizes worst-case rationale, specific analytical methods, and validated recovery. For medical device manufacturers, cleaning validation usually concerns manufacturing residuals and, where relevant, reprocessing, under the Quality Management System Regulation, which took effect February 2, 2026 and incorporates ISO 13485:2016 by reference. The residue toxicology logic is the same even when the regulatory citation differs.
The point to carry forward: a limit defended only by "1/1000th of a dose" or "10 ppm" with no toxicological basis is the framing inspectors now question. Anchor to a PDE or ADE.
Selecting the worst case
You cannot validate cleaning for every product and every location on every piece of equipment, and regulators do not expect you to. They expect a defensible worst-case selection so that validating the hardest scenario justifies the easier ones. Worst case operates on three axes.
Worst-case product is the product in a shared-equipment group that is hardest to clean and most hazardous to carry over. Two factors combine: cleanability, which reflects solubility, stickiness, and how the residue behaves after drying, and toxicity or potency, which reflects how small a carryover amount becomes a problem. A poorly soluble, highly potent compound is the classic worst case. Where one product is not clearly worst on both factors, some programs validate the hardest-to-clean product and the most-potent product separately rather than force a single choice.
Worst-case location is the hardest-to-clean surface on the equipment: dead legs, valve seats, gaskets, agitator shafts, welds, and any geometry that traps residue or resists the cleaning agent. Sampling locations should be chosen because they are hard to clean, not because they are easy to reach. Choosing convenient locations and calling them representative is one of the most common findings.
Worst-case condition covers the operational extremes the cleaning procedure has to handle: the maximum dirty hold time before cleaning starts, the largest campaign length before a full clean, and the most concentrated or most degraded residue state. Validating at the benign end of these conditions and running production at the harsh end is an obvious gap.
Grouping, also called bracketing or the family approach, is how worst case scales. Products and equipment are grouped by similarity, and the worst case within each group represents the group. Grouping is legitimate and expected, but the grouping rationale has to be documented and the group boundaries have to make sense. A group that lumps a freely soluble low-potency product with an insoluble high-potency one because they run on the same line is a group an inspector will take apart.
Setting the carryover limit
The maximum allowable carryover, or MACO, is the largest amount of the previous product that may remain and still be safe in the next product. The health-based calculation starts from the PDE.
Conceptually, the MACO is the PDE of the previous product scaled by how much of the next product a patient takes and how large the next batch is. In words rather than a single formula: take the safe daily exposure to the previous compound, divide by the largest daily intake of the next product, and multiply by the batch size of the next product. The result is the total amount of previous product that may be spread across an entire next batch without any patient exceeding the safe exposure. The MACO is then divided across the shared product-contact surface area to give a limit per unit area, which is what a swab result is compared against, and can also be expressed as a concentration in the rinse.
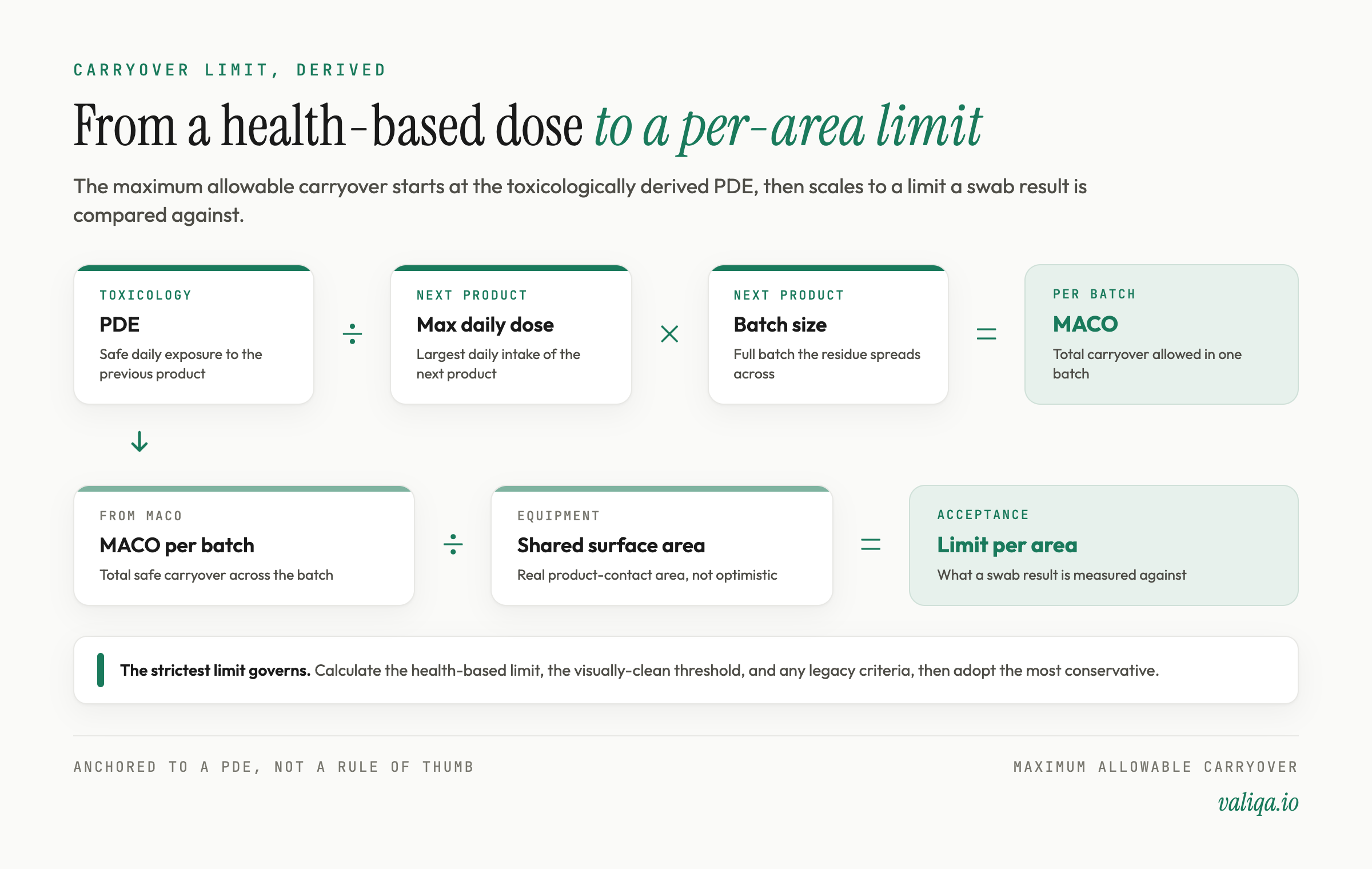
Two practical points decide whether this holds up. First, the PDE must come from a documented toxicological evaluation by a qualified person, not from a number copied between spreadsheets. Second, when you compute a per-area limit you need the real shared surface area and a realistic sampled area, because an optimistic surface-area figure quietly inflates the apparent margin. Where several limit approaches are available, the conservative one governs: many programs calculate the health-based limit, the visually-clean threshold, and any legacy criteria, and adopt the strictest.
Visually clean is a necessary condition, not a sufficient one. Every cleaned surface should be visually free of residue, and the visible-residue threshold should itself be established. But visually clean does not prove the surface is below the health-based limit for a potent compound, so it supplements the analytical limit rather than replacing it.
Swab versus rinse sampling
The two sampling methods answer different questions, and mature programs usually use both.
Swab sampling physically wipes a defined area with a moistened swab, which is then extracted and analyzed. Its strength is direct access to worst-case locations: you can swab the specific dead leg or gasket that is hardest to clean. Its limitations are that it only covers the areas you reach, it depends heavily on consistent technique, and it cannot reach the interior of long transfer lines or complex geometries. Swabbing is the method of choice for identifiable worst-case surfaces.
Rinse sampling collects a final rinse of the equipment and analyzes it, giving an averaged picture across all wetted surfaces including places a swab cannot reach. Its strength is coverage of large or inaccessible surface area. Its weakness is dilution and averaging: a localized hot spot can be masked by clean surface everywhere else, and residue that is not readily rinsed off may not show up. Rinse sampling is the method of choice for large surface areas and hard-to-access internal geometry.
Because each method is blind to what the other catches, using swab sampling on the accessible worst-case locations and rinse sampling for overall coverage is the combination that tends to satisfy reviewers. Whatever the choice, the protocol has to justify why the method suits the equipment, not just state which method was used.

Recovery studies underpin every result
A sampling result is only as trustworthy as the recovery study behind it. No swab or rinse recovers 100 percent of the residue on a surface, so a recovery study spikes a known amount of residue onto a representative coupon of the actual surface material, samples it by the intended method, and measures the percentage recovered. Every reported cleaning result is then corrected by that recovery factor.
Recovery studies matter more than teams expect, and skipping them is a frequent finding. The recovery factor depends on the surface material, so stainless steel, PTFE, and glass each need their own study. It depends on the residue, so different products need their own recovery data. And a very low recovery factor is itself a warning: if you only recover a small fraction of what is on the surface, your method is not reliable enough to prove cleanliness, and the fix is a better sampling method, not a generous correction applied to a weak one. Regulators expect recovery to be established and applied, and a validation result reported without recovery correction is incomplete.
Justifying hold times
Two hold times bound the cleaning process, and both need their own supporting data.
Dirty hold time is the maximum time equipment may sit soiled between the end of production and the start of cleaning. As residue dries and ages it becomes harder to remove and, on wet surfaces, microbial load grows. The dirty hold time you validate has to be at least as long as the worst case production actually allows, and the validation runs should be executed at that maximum, not at a convenient short interval.
Clean hold time is the maximum time cleaned equipment may sit before it must be used or re-cleaned, and it is primarily a microbial and recontamination concern. Establishing it means holding cleaned equipment for the claimed duration under representative storage conditions and demonstrating that bioburden and, where relevant, endotoxin stay within limits.
Both hold times are acceptance criteria, not footnotes. A program that validates cleaning but never establishes the hold times, then routinely holds equipment dirty overnight or clean for a week, has a real gap between what was validated and what is practiced. That gap between the validated condition and the operating condition is a recurring theme across validation, and it is the same reasoning that drives revalidation when equipment or processes change.
Where cleaning validation is commonly flagged
The findings cluster in predictable places. Sampling locations chosen for accessibility rather than difficulty. Carryover limits with no toxicological basis, still resting on legacy rules of thumb alone. Recovery studies missing, or applied to the wrong surface material. Worst-case grouping with no documented rationale, or groups that mix products that should not share a bracket. Hold times not validated, or validated shorter than production actually runs. And the quiet one: a validated procedure that has drifted from the SOP operators actually follow, so the evidence describes a process that is no longer performed.
Each of these traces back to the same root cause: a break in the chain from limit to rationale to result. Cleaning validation is defensible when every number has a reason behind it that an inspector can follow, from the toxicological limit through the worst-case selection and the recovery-corrected sampling result. When that chain is intact, the program holds. When any link is asserted rather than justified, that is where the observation lands.
Building the evidence chain
Cleaning validation is not a single document, it is a connected set: the risk assessment and grouping rationale, the toxicological evaluation behind the limits, the protocol with worst-case selection and acceptance criteria, the recovery studies, and the executed results with deviations and dispositions. Each piece references the next, and an auditor can start anywhere and walk the chain in either direction.
That connectedness is exactly what a validation platform is meant to hold, so that the worst-case rationale, the limit basis, the sampling plan, and the executed evidence live together with traceability rather than scattered across spreadsheets and email. If you are structuring a cleaning validation program alongside your equipment qualification work, the full IQ/OQ/PQ lifecycle guide covers how the surrounding qualification pieces fit together.
Frequently Asked Questions
Ready to automate your validation documentation?
Generate audit-ready IQ/OQ/PQ protocols in minutes, not weeks.
Get StartedWe use essential cookies for authentication and security. With your consent, we also use Microsoft Clarity on our marketing pages to understand how visitors navigate the site. Learn more.