PPQ Protocol Guide: Process Performance Qualification Step by Step
By the time a process reaches performance qualification, the easy questions have been answered. The equipment was installed correctly and the installation qualification proved it. The equipment runs within its specified ranges and the operational qualification proved that too. What nobody has proven yet is the thing that actually matters to a patient: that this facility, with this equipment, these operators, these materials, and this exact set of process parameters, will produce a good product run after run without anyone hovering over it. That is the question process performance qualification exists to answer, and the PPQ protocol is where you write down how you intend to answer it.
PPQ is where many otherwise solid validation efforts get vague. Teams that write crisp IQ and OQ protocols suddenly reach for boilerplate: "three successful batches," a sampling plan copied from the last product, acceptance criteria that restate the specification without explaining why the process can meet it. This guide is for the engineer who has to write the PPQ protocol properly: where PPQ sits in the modern process validation lifecycle, how it differs from the equipment qualification that came before it, what the protocol has to contain section by section, how to size the number of runs and the sampling without inventing numbers, and how to write the whole thing so it reads as evidence rather than ritual.
Where PPQ sits in the validation lifecycle
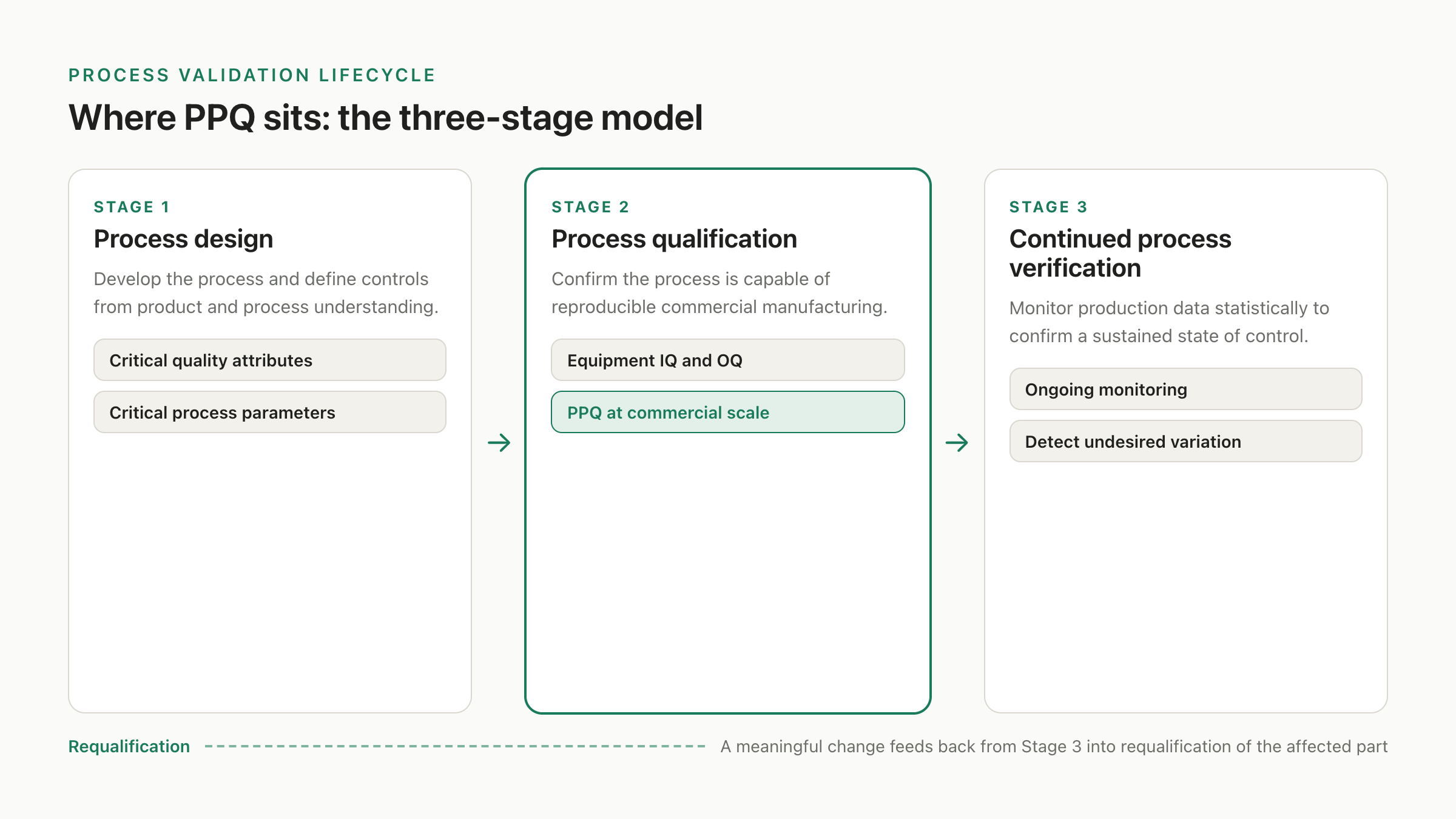
Process performance qualification is a stage in a larger model, and the protocol only makes sense if you know which stage. The FDA's 2011 guidance, "Process Validation: General Principles and Practices," frames process validation as a lifecycle in three stages. Stage 1 is process design, where you develop the process and define the controls based on product and process understanding. Stage 2 is process qualification, where you confirm the process is capable of reproducible commercial manufacturing. Stage 3 is continued process verification, where ongoing statistical monitoring of production data confirms the process stays in a state of control and surfaces undesired variation across its commercial life.
PPQ lives inside Stage 2. Stage 2 actually has two parts: the qualification of the facility, utilities, and equipment, including the installation and operational qualification of that equipment, and then the process performance qualification itself, which combines all of those qualified elements with the trained personnel, the verified materials, the commercial process, and the control procedures to actually manufacture commercial batches under a protocol. PPQ is the culmination of Stage 2. It is the point where you stop testing components in isolation and run the whole integrated process at commercial scale, under heightened scrutiny, to demonstrate that the design from Stage 1 holds up in the real plant.
Two consequences follow from that placement. First, PPQ has prerequisites that are not optional: the equipment qualification has to be complete and approved, the analytical methods have to be validated, the operators have to be trained, and the materials have to be from qualified sources, because PPQ assumes all of those are already in a state of control and tests only the process built on top of them. Second, PPQ is not the end. A successful PPQ releases the process into routine production, but Stage 3 then watches it continuously, and a meaningful change later can trigger requalification of the affected part. PPQ is a milestone in a lifecycle, not a one-time certificate.
PPQ versus equipment PQ: the distinction that trips people up
The single most common source of confusion is the letter Q. In medical device manufacturing, the process validation sequence is usually written IQ, OQ, PQ, where PQ stands for performance qualification and is the third stage that demonstrates the process consistently produces conforming product under anticipated conditions. That IQ, OQ, PQ framework is recognized industry convention with roots in the GHTF process validation guidance rather than a structure any single regulation names. The requirement it serves is the regulation: for medical devices under FDA jurisdiction, process validation now lives in ISO 13485:2016 clause 7.5.6, incorporated by reference into 21 CFR Part 820 when the Quality Management System Regulation took effect on February 2, 2026, and expressed before that at 21 CFR 820.75. In the pharmaceutical lifecycle language of the 2011 guidance, the equivalent commercial-scale demonstration is called PPQ, process performance qualification. They are answering the same fundamental question, but the vocabulary comes from different regulatory traditions, and a protocol that mixes them carelessly invites confusion.
There is also a narrower technical distinction worth keeping straight even within one vocabulary. An equipment performance qualification asks whether a given piece of equipment performs to its requirements under loaded, real-use conditions. Process performance qualification asks whether the entire process, of which that equipment is one part, delivers a conforming product. A qualified autoclave can pass its own performance qualification and still sit inside a sterilization process that has not been process-qualified, because the process wraps in loading patterns, cycle selection, bioburden, and product configuration that the equipment qualification never touched. The IQ, OQ, and PQ breakdown covers where that line falls in more detail; for the PPQ protocol, the rule is simply to state explicitly which scope you are qualifying so no reader has to guess whether "PQ" meant the machine or the process.
For this guide, PPQ means the commercial-scale process demonstration, whichever letter your industry stamps on it. The structure below applies either way.
What the process design stage owes the protocol
A PPQ protocol cannot be written from scratch at the qualification stage, and a protocol that tries to is usually the one full of borrowed boilerplate. PPQ inherits its content from Stage 1. The process design work is where you identified the critical quality attributes, the product properties that have to be controlled to keep the product safe and effective, and the critical process parameters, the inputs whose variation affects those attributes. It is also where you established the normal operating ranges and the proven acceptable ranges for those parameters through development studies and process characterization experiments. Risk assessment points you at which parameters are worth characterizing; the ranges themselves come from the data those studies generate.
That risk assessment is the spine of the protocol. The same process risk analysis you would run in a PFMEA is what tells you which parameters are critical, which attributes are at risk, and therefore what the PPQ has to watch most closely. If a sealing temperature drives seal integrity and seal integrity is a critical quality attribute, the PPQ protocol has to demonstrate the process holds that temperature in its acceptable range and produces seals that pass, and it has to sample enough seals to mean something. PPQ does not rediscover what is critical. It inherits that list from process design and proves the commercial process controls it. If your protocol cannot point back to where each parameter and attribute came from, the process design stage was either incomplete or undocumented, and that gap will surface in the protocol as arbitrary-looking criteria.
The PPQ protocol, section by section
A PPQ protocol is a controlled document with a predictable spine. The sections below are the ones that carry the weight. The FDA's 2011 guidance does not mandate a rigid heading list, but it does describe the content a PPQ protocol should address, and these sections cover it.
Purpose and scope. State the process being qualified, the product or product family, the facility and line, the batch or lot scale, and the boundaries of what this protocol covers. Name what is explicitly out of scope, such as upstream processes qualified separately. This is also where you state which qualification model you are using and what "PPQ" means in your terminology, so the rest of the document is unambiguous.
Process description. Describe the commercial manufacturing process step by step, the equipment used at each step, the in-process controls, and where the critical process parameters and critical quality attributes sit along the flow. An auditor reads this to understand what should have been monitored and where.
Prerequisites and entry criteria. The conditions that must be true before the first PPQ run begins, each with a reference to its evidence: equipment IQ and OQ complete and approved, utilities qualified, analytical methods validated, cleaning validation status, operators trained and qualified, materials sourced from qualified suppliers, and the master batch record or device manufacturing record approved. PPQ assumes a qualified foundation; this section is where you prove the foundation is there rather than asserting it.
Critical quality attributes and critical process parameters. The table that ties the protocol back to process design. List each critical quality attribute with its specification, and each critical process parameter with its normal operating range and proven acceptable range, and reference the risk assessment and development data that established them. Everything the protocol tests should trace to a row here.
Manufacturing conditions. The actual parameter set points and ranges the process will run at during PPQ, the equipment settings, the in-process controls and their limits, and any conditions you are deliberately challenging. PPQ generally runs at the target commercial conditions rather than at the edges; the edges were explored during OQ and process design. State that choice and its rationale.
Sampling plan. Where you sample, when, how often, and how many, for both in-process and finished attributes. PPQ uses heightened sampling, more than routine production will, because the point is to characterize the process with statistical confidence before you trust it. The plan has to say not just the numbers but the justification: why this many samples from these locations gives you the confidence you are claiming. More on sizing this below.
Tests and acceptance criteria. For each attribute, the test method, the acceptance criterion, and how results will be judged. Write acceptance criteria that are specific and defensible, tied to the product specification and to statistical expectations where appropriate, not vague statements like "results meet requirements." A criterion of "all individual fill weights within label claim limits and the run mean within the validated range with a stated confidence" is testable; "fill weight acceptable" is not.
Number of runs and statistical rationale. How many batches or lots you will run and why. This is the section teams most often fill with a reflexive number; it deserves a real justification, covered in its own section below.
Data collection and analysis. What data is recorded, how, and the statistical methods you will use to evaluate it, such as the calculation of process capability or the analysis of variation within and between batches. Decide the analysis before you run, not after you see the results.
Deviation handling. How a deviation or an out-of-specification result during PPQ is documented, investigated, and dispositioned, and what it means for the validation conclusion. A deviation during PPQ is not automatically a failure, but it has to be assessed honestly against its impact, and the protocol has to say how.
Roles, responsibilities, and approval. Who executes, who reviews, who owns the process, and the quality unit approval that the 2011 guidance expects on both the protocol and the final report. PPQ protocols and reports are reviewed and approved by the appropriate functions including quality assurance.
Conclusion criteria. The explicit statement of what outcome lets you conclude the process is validated and release it to routine production, written before execution so the conclusion is not negotiated after the data is in.

How many runs, and why "three batches" is no longer the answer
For decades the reflexive answer to "how many PPQ batches" was three. Three consecutive successful batches became shorthand for a validated process, and you can still hear it in plants today. The 2011 guidance deliberately moved away from a fixed number. It states that the number of PPQ batches should be sufficient to provide statistical confidence that the process is reproducible and will consistently deliver quality product, and that the appropriate number depends on the variability of the process, the complexity of the product, process and product experience, and the overall level of process understanding gained during development. There is no magic number, and citing one as if a regulation requires it is itself a finding waiting to happen.
What replaces the number is a justification. The right number of runs is the number that gives you the confidence your conclusion claims, given how variable and how well understood the process is. A simple, well characterized process with a long development history and tight inherent variability may justify fewer runs with heavy sampling within each. A complex or novel process with less prior data justifies more. Some organizations adopt a continuous or hybrid approach, qualifying with an initial set of runs and continuing enhanced verification into early commercial production until the data supports normal monitoring. Whatever you choose, the protocol has to state the number and the reasoning, grounded in the variability, complexity, and process understanding behind it, not in habit. Three may well be the answer you land on. It just cannot be the answer you start with.
Sizing the sampling so it means something
The number of runs is only half of the statistical story; the sampling within each run is the other half. PPQ sampling is heavier than routine sampling on purpose. During routine production you sample enough to maintain control of a process you already trust. During PPQ you sample enough to earn that trust, which means more samples, from more locations, at more time points, chosen to expose variation rather than to confirm a happy result.
Good PPQ sampling is designed around the sources of variation the process can actually have. Within a single batch, that might mean sampling at the beginning, middle, and end of a run to catch drift, and across the width or the cavities of a multi-station tool to catch position effects. Across batches, it means enough lots to characterize batch-to-batch variation, not just within-batch. The sampling plan should be tied to a statistical basis: a stated confidence and reliability, a sampling approach appropriate to the attribute, and a sample size that supports the claim you intend to make in the conclusion. The cardinal sin is the round number with no rationale, twenty samples because twenty feels thorough. A reviewer can reasonably ask why twenty and not ten or fifty, and "it seemed enough" is not an answer. The protocol has to connect the sample size to the confidence it buys.
A worked example: qualifying a liquid fill process
Take a process that fills a liquid drug product into vials on a commercial filling line. Stage 1 development identified the critical quality attributes as fill volume, container closure integrity, and sterility, and the critical process parameters as fill pump settings, line speed, and stopper placement force, each with an established normal operating range and a proven acceptable range from development and OQ studies. The equipment is qualified: the filler passed its IQ and OQ, the sterilizing and depyrogenation steps are qualified, the environmental controls are qualified, the fill-weight analytical method is validated, and the operators are trained. Those facts are the prerequisites section, each with a reference.
The PPQ protocol runs the process at its target commercial conditions, not at the edges, because the edges were already explored. The sampling plan calls for fill-volume checks at defined intervals across each run, including start, middle, and end, plus samples spanning the relevant positions of the filling heads, with a sample size justified to detect a meaningful shift in mean or variability at a stated confidence. Container closure integrity and the relevant sterility assurance attributes are sampled per their own statistically based plans. The acceptance criteria state, per attribute, the specification limit and the statistical expectation, for example that all sampled fill volumes fall within limits and the run statistics support the claimed capability. The number of runs is justified from the process variability and the depth of development understanding, and that justification is written into the protocol, not assumed.
When the runs execute, the data is captured and analyzed by the predefined methods. If a fill-volume excursion occurs, it is logged as a deviation, investigated for cause and impact, and dispositioned, and its effect on the conclusion is assessed honestly rather than waved away. If every attribute meets its criteria across the runs and the statistics support reproducibility, the conclusion section, written before execution, lets the team declare the process validated and release it to routine production under Stage 3 monitoring. Every number in that protocol traces to something: the parameters to process design, the sampling to a statistical basis, the acceptance criteria to the specification, and the run count to the process understanding behind it.
Common ways a PPQ protocol gets flagged
The first is starting from a batch count instead of a question. A protocol that opens with "three batches will be run" and reverse-engineers the rest has the logic backward. The number of runs is a conclusion from the variability and process understanding, and a protocol that cannot show that reasoning reads as ritual.
The second is acceptance criteria that merely restate the specification without a statistical dimension. PPQ is supposed to demonstrate reproducibility and capability, not just that one batch happened to pass. Criteria that ignore variation and confidence leave the central PPQ claim unproven.
The third is treating PPQ as independent of everything before it. A protocol that does not establish its prerequisites, or that tests parameters with no traceable link to process design and risk assessment, is qualifying a process whose foundation is unverified. The strength of a PPQ protocol comes from the qualified equipment, validated methods, and documented design knowledge it rests on, the same way a validation master plan ties the whole program together.
The fourth is sampling plans with numbers but no rationale, and conclusion criteria written after the data is in. Both turn a scientific exercise into a paperwork one. The fix for each is the same discipline that keeps any protocol defensible: decide the criteria and the analysis before execution, and connect every number to the confidence it is supposed to provide.
Where this leaves the engineer
A PPQ protocol is not a heavier OQ and it is not a stack of three batch records with a cover page. It is the document where you commit, in advance, to how you will prove that an integrated commercial process delivers conforming product reproducibly, and then let the data either support that conclusion or not. Place it correctly in the lifecycle, as the culmination of Stage 2 resting on qualified equipment and a documented design. Inherit your critical attributes and parameters from process design rather than reinventing them. Size the runs and the sampling from variability and confidence rather than habit. Write acceptance criteria and conclusion criteria before you run, with the statistics built in. Handle deviations honestly. Do that, and the PPQ stops being the vague stage at the end of qualification and becomes what it is meant to be: the moment a process earns the right to be trusted in routine production.
---
Valiqa is an AI-powered validation lifecycle platform for regulated manufacturing. Learn more at valiqa.io
Frequently Asked Questions
Ready to automate your validation documentation?
Generate audit-ready IQ/OQ/PQ protocols in minutes, not weeks.
Get StartedWe use essential cookies for authentication and security. With your consent, we also use Microsoft Clarity on our marketing pages to understand how visitors navigate the site. Learn more.