Design Qualification (DQ): What It Is, When You Need It, and How to Write One
Most validation teams can recite IQ, OQ, and PQ in their sleep. Ask the same team where Design Qualification fits and the answers scatter. Some skip it entirely. Some run it after the equipment is already on the floor, which defeats the point. Some produce a two-page document that restates the purchase order and call it done. All three are common, and all three leave a gap an auditor can walk through.
Design Qualification, or DQ, is the stage that checks the design of a system against what you actually need before you spend money building or buying it. Done well, it is the cheapest place in the entire validation lifecycle to catch a problem. Done late or not at all, the same problem surfaces during IQ or OQ, when fixing it means change orders, re-tests, and schedule slip.
This guide covers what DQ verifies, how it differs from IQ, when it is worth the effort and when it is not, the sections a defensible DQ protocol contains, and how it fits under current US and EU frameworks.
What Design Qualification actually verifies
EU GMP Annex 15 gives the cleanest definition. Design Qualification is the documented verification that the proposed design of the facilities, systems, and equipment is suitable for the intended purpose. The operative words are proposed design and intended purpose. DQ happens while the system is still a specification, a drawing, or a vendor proposal, and it confirms that this design will meet the requirements you wrote down.
Those requirements live in the User Requirements Specification, or URS. The URS states what the system must do: the process parameters it must hit, the capacity it must handle, the materials of construction, the data integrity and Part 11 controls it must support, the utilities it needs, the regulatory expectations it must satisfy. DQ is the checkpoint where you compare the vendor design or engineering specification back to that URS, line by line, and document that every requirement is addressed by the design.
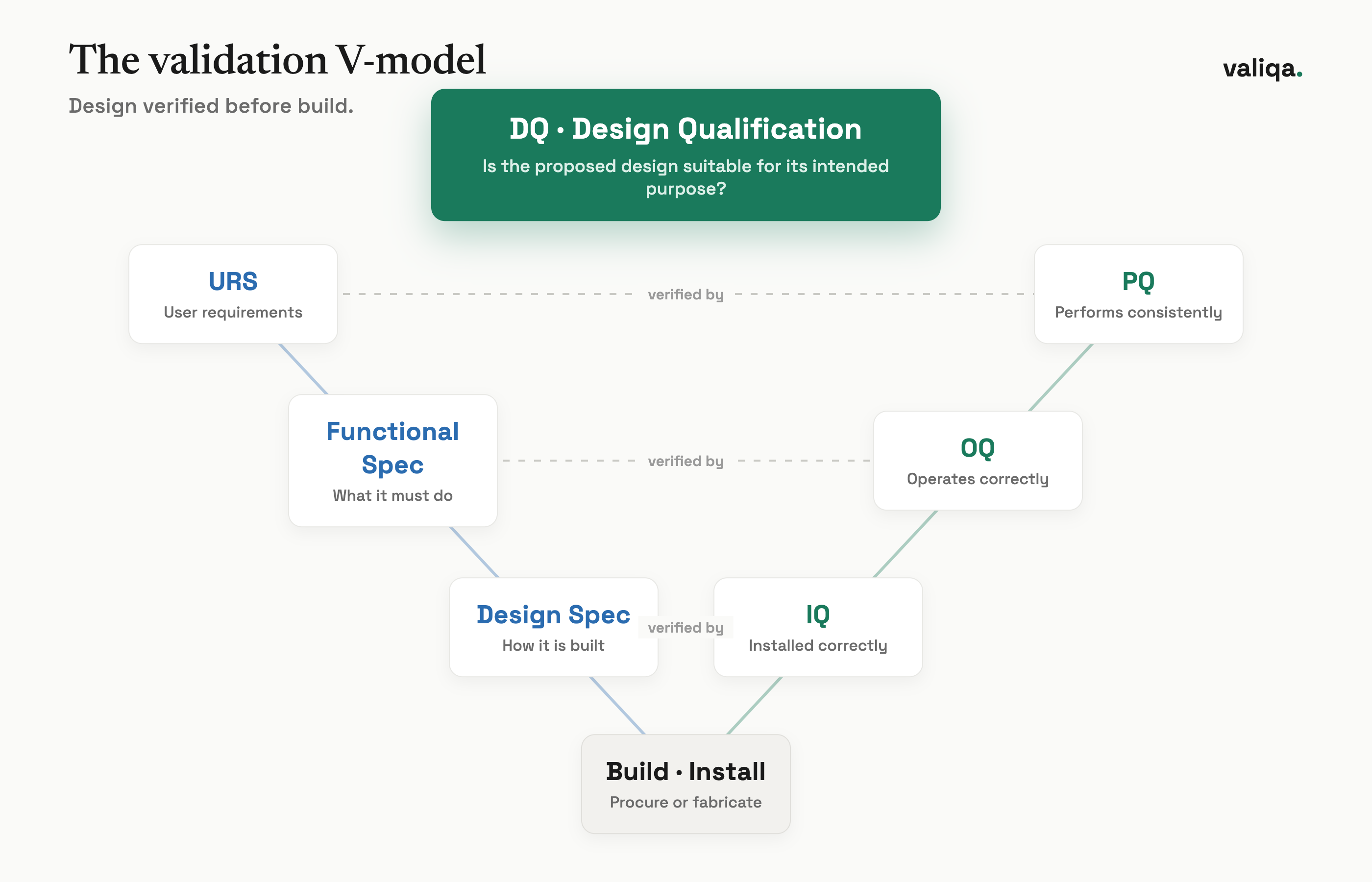
The V-model above is the simplest way to hold the relationship in your head. The left side descends from user requirements down through functional and design specifications. The right side climbs back up through IQ, OQ, and PQ. DQ sits at the top of the descent. It closes the loop between what you asked for and what the design promises to deliver, before anyone builds anything. IQ, OQ, and PQ then verify that the delivered system matches the design and performs. If DQ is missing, the right side of the V has nothing solid to verify against, because no one confirmed the design was correct in the first place.
DQ versus IQ: the confusion worth clearing up
The most common mix-up is treating DQ and Installation Qualification as the same activity at different times. They are different questions.
DQ asks: is this the right design? It is a paper exercise performed on specifications, drawings, P&IDs, and vendor documentation. Nothing is installed. You are checking suitability of intent.
IQ asks: was the right thing installed correctly? It is a physical verification performed on the delivered equipment. You confirm the unit matches the purchase specification, the utilities are connected, the calibration certificates are present, the materials of construction match what was specified, and the documentation set is complete.
A concrete example. Your URS says a jacketed vessel must hold a product contact surface finish of 20 Ra or better because of a cleaning requirement. DQ is where you read the vendor drawing, confirm the specified finish is 20 Ra or better, and document that the design meets the requirement. IQ is where the vessel arrives and you verify the actual delivered surface finish certificate reads 20 Ra or better. DQ caught whether the vendor even promised the right finish. IQ caught whether they delivered it. Skip DQ and you can install a beautifully documented vessel with the wrong finish, because no one checked the promise against the requirement.
When you actually need DQ, and when you do not
DQ is not free, and it is not always warranted. The scope of DQ should be risk-based and proportionate to how custom and how critical the system is. This is the same risk logic that decides whether a piece of equipment needs IQ only or the full IQ, OQ, PQ sequence.
DQ earns its place when the design is not a known quantity:
Custom or engineered-to-order equipment. If a vendor is building something to your specification, DQ is the only stage that catches a design that does not match the URS before fabrication. This is where DQ pays for itself most clearly.
Complex or integrated systems. A packaging line, a filling and capping system, a water-for-injection skid, or any system with meaningful automation carries design risk that a purchase order cannot capture.
Computerized systems in the higher GAMP categories. For configured or custom software, GAMP 5 folds design suitability into the specification and verification approach, and the design review is where you confirm the system architecture, data integrity controls, and Part 11 support match requirements. If you are unsure which category applies, the GAMP 5 categories guide walks through the distinctions.
Facilities and utilities. Cleanroom design, HVAC classification, and utility systems are expensive to change once built, which makes the design-stage checkpoint especially valuable.
DQ is usually not warranted for simple, standard, off-the-shelf equipment used within its normal operating range: a calibrated balance, a standard pH meter, a general-purpose oven with no custom configuration. For those, a well-scoped IQ and OQ carry the load, and a formal DQ adds paper without adding assurance. The judgment call is whether the design carries risk that a specification and an IQ would not otherwise surface. If the vendor catalog fully defines the item and it meets your URS on its face, a documented specification review inside the purchasing process is often proportionate, and a standalone DQ protocol is overkill.
The failure mode to avoid is the reflex to either DQ everything or DQ nothing. Both are hard to defend. A validation program that qualifies design effort against risk, and records the rationale, is the defensible position, and it is exactly the kind of decision your Validation Master Plan should govern.
What goes into a DQ protocol
A DQ protocol is short compared to an OQ, but it has a definite structure. The sections below are the ones a reviewer expects to see.
Purpose and scope. State the system under qualification, the boundaries of the design being assessed, and what is explicitly out of scope. Name the URS revision and the design documents under review by document number and revision, because a DQ against an uncontrolled draft specification is worth very little.
References. The URS, the functional specification and design specification if they exist, applicable standards, the risk assessment, and the governing VMP. DQ is a comparison exercise, so the documents being compared have to be identified precisely.
Regulatory and GMP design review. Confirm that the design supports the regulatory expectations that apply: cleanability and material compatibility for product contact surfaces, data integrity and access control for computerized elements, environmental and utility requirements, and safety. This is where a design that is functionally fine but not GMP-suitable gets caught.
Supplier assessment linkage. DQ is the natural place to reference the outcome of vendor qualification and any Factory Acceptance Test planning. A design review is more credible when the supplier building it has been assessed and the design documents come from a controlled source.
The design-to-URS verification. This is the core of the DQ and it is a traceability exercise. Every URS requirement is listed, mapped to the design element that satisfies it, and given a documented verdict. The requirement for a 20 Ra product contact finish maps to a line on the vendor drawing. The requirement for audit-trail retention maps to a functional specification section. The requirement for a specific throughput maps to the engineering calculation or vendor performance data. Each row gets a result and a reference.
Acceptance criteria. For DQ, the acceptance criteria are usually pass or fail against each traced requirement: the design either addresses the requirement or it does not, with the evidence cited. Write them the same way you would write any acceptance criteria that will survive an audit, anchored to the URS and objective rather than to opinion.
Deviations. Any requirement the design does not meet is a DQ deviation, documented with its disposition: change the design, change the requirement with justification, or accept with a documented risk rationale. A DQ with zero deviations on a custom system is often a sign the review was shallow, not that the design was perfect.
Conclusion and approval. A statement that the design is or is not suitable for intended use, and the signatures that make it a controlled record. DQ is typically owned by engineering or validation and approved by quality, the same ownership split that runs through the rest of the qualification lifecycle.
The traceability thread that DQ starts
The most valuable thing a good DQ produces is not the pass or fail verdict. It is the first link in a traceability chain that runs through the entire validation lifecycle. The URS requirement traced in DQ becomes the design element verified in IQ, the function tested in OQ, and the performance demonstrated in PQ. When an auditor asks how you know a specific requirement was met, a clean chain answers in one line: URS requirement to design element to installation check to functional test to performance run to summary report.
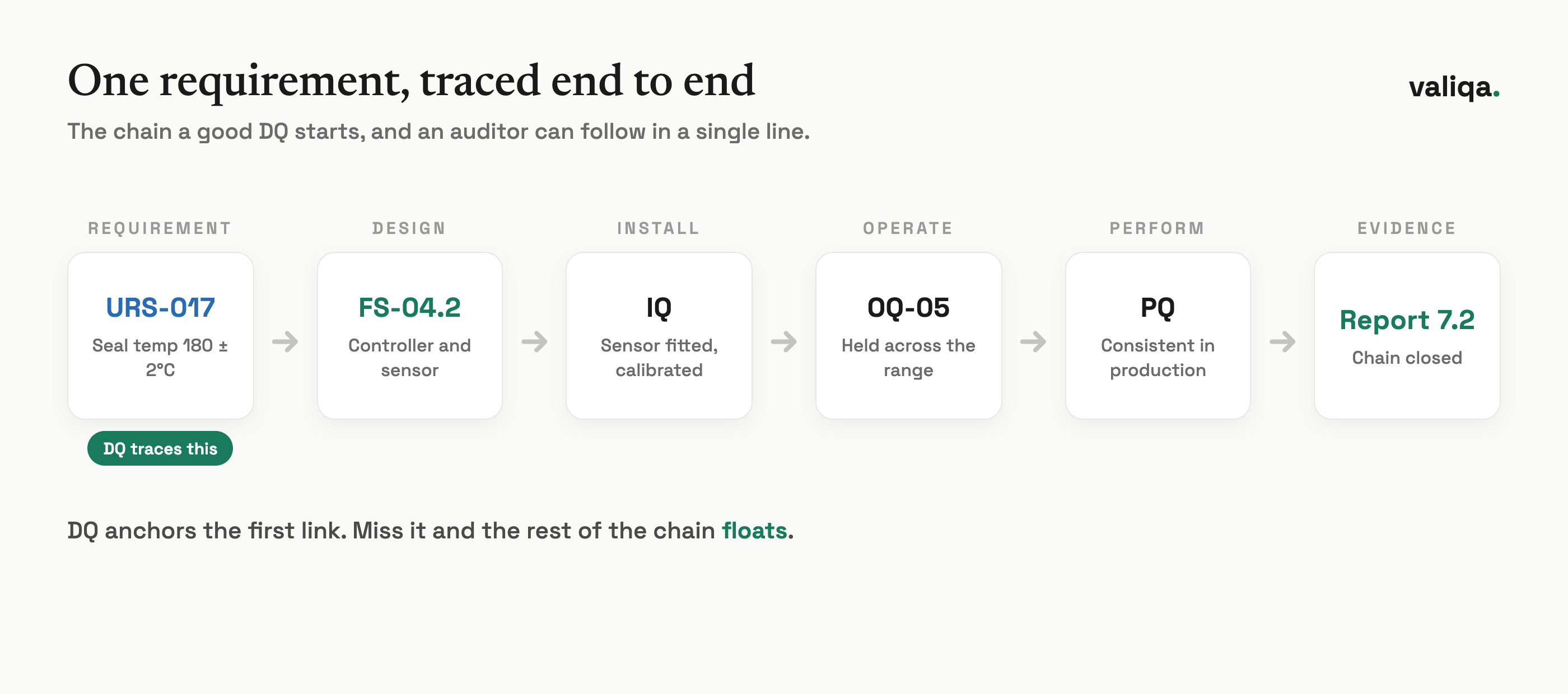
DQ is where that chain gets its first anchor. If the URS requirement is never traced to a design element, the rest of the chain floats. This is why a DQ that is really just a restated purchase order is a missed opportunity: it produces a signature without producing traceability, and traceability is the thing that makes the whole qualification program defensible.
A short worked example
A device manufacturer is buying a custom heat sealer for a sterile barrier system. The URS specifies a sealing temperature range of 140 to 180 degrees C, a dwell time adjustable from 0.5 to 3.0 seconds, seal pressure repeatable within a stated tolerance, an audit trail on all parameter changes, and product contact tooling in a specified alloy.
At DQ, the team reviews the vendor design package against those requirements. Five of six requirements trace cleanly to the drawing and functional specification. The sixth, the audit trail, reveals that the vendor control system logs parameter changes but does not attribute them to a unique user, which the URS requires for Part 11 alignment. That is a DQ deviation. Because it is caught at the design stage, the fix is a configuration change and a firmware option the vendor adds before shipping, at no schedule cost. Had the same gap surfaced at OQ, the sealer would already be installed, the OQ would fail, and the fix would mean a return visit, a re-test, and a documented deviation on a system already in the validation record. The DQ turned a change order into a checkbox.
Where DQ fits under current frameworks
DQ is most explicitly named in EU GMP Annex 15, which lists Design Qualification as the first element of equipment and facility qualification and defines it as the documented verification that the design is suitable for intended purpose. For computerized systems, GAMP 5 Second Edition treats design suitability through its specification and verification framework, scaled to the system category.
In the US framework the word DQ appears less often, but the activity does not disappear. The FDA process validation guidance describes Stage 1, Process Design, which includes designing a process and the equipment to support it so that it is capable of consistently producing quality product. Verifying that an equipment design supports the intended process is the same checkpoint DQ performs. For medical device manufacturers, the design of the finished device is governed by design controls under ISO 13485:2016 Section 7.3, now the anchor for US requirements through the Quality Management System Regulation that took effect on February 2, 2026, incorporating ISO 13485:2016 by reference. Equipment DQ is distinct from finished-device design controls, but it supports process validation under ISO 13485:2016 Section 7.5.6 and connects to purchasing controls, because qualifying the design of the equipment that makes the product is part of demonstrating the process is capable.
The practical read across all of these: whether or not your regulatory framework uses the letters DQ, verifying that a design meets requirements before you build it is expected engineering discipline, and documenting that verification is what turns discipline into evidence.
How Valiqa fits
Valiqa generates DQ protocols alongside the rest of the qualification lifecycle, grounded in the equipment details and requirements you provide. The generated DQ carries the URS-to-design traceability structure into the IQ, OQ, and PQ protocols that follow, so the chain that starts at design qualification stays intact through to the summary report. The engineer still owns the review and the judgment about what the design must satisfy. The tool removes the blank-page work of structuring the protocol and keeps the traceability consistent across every stage.
Design Qualification is not the most visible stage in the validation lifecycle, but it is the one with the best return on effort. It catches design problems at the only point where they are cheap to fix, and it plants the traceability that makes everything downstream defensible. Skip it on a custom system and you are not saving time, you are moving the cost to a more expensive place.
---
Valiqa is an AI-powered validation lifecycle platform for regulated manufacturing. Learn more at valiqa.io
Frequently Asked Questions
Ready to automate your validation documentation?
Generate audit-ready IQ/OQ/PQ protocols in minutes, not weeks.
Get StartedWe use essential cookies for authentication and security. With your consent, we also use Microsoft Clarity on our marketing pages to understand how visitors navigate the site. Learn more.